Co-occurrence of frameshift mutations in smad6 and tcf12 in a child with complex craniosynostosis
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Non-syndromic craniosynostosis (CS) affects 1 in 2350 live births. Recent studies have shown that a significant fraction of cases are caused by de novo or rare transmitted mutations
that promote premature osteoblast differentiation in cranial sutures. Rare heterozygous loss-of-function (LOF) mutations in _SMAD6_ and _TCF12_ are highly enriched in patients with
non-syndromic sagittal and coronal CS, respectively. Interestingly, both mutations show striking incomplete penetrance, suggesting a role for modifying alleles; in the case of _SMAD6_, a
common variant near _BMP2_ drastically increases penetrance of sagittal CS. Here, we report a proband presenting with both sagittal and coronal craniosynostosis with the highly unusual
recurrence of CS within two months of initial surgery, requiring a second operation to re-establish suture patency at six months of age. Exome sequencing revealed a rare transmitted
frameshift mutation in _SMAD6_ (p. 152 fs*27) inherited from an unaffected parent, absence of the common _BMP2_ risk variant, and a de novo frameshift mutation in _TCF12_ (p.E548fs*14).
_SMAD6_ and _TCF12_ independently inhibit transcriptional targets of BMP signaling. The findings are consistent with epistasis of these mutations, increasing penetrance and severity of CS in
this proband. They also add to the list of composite phenotypes resulting from two Mendelian mutations, and support the utility of exome sequencing in atypical CS cases. The female proband
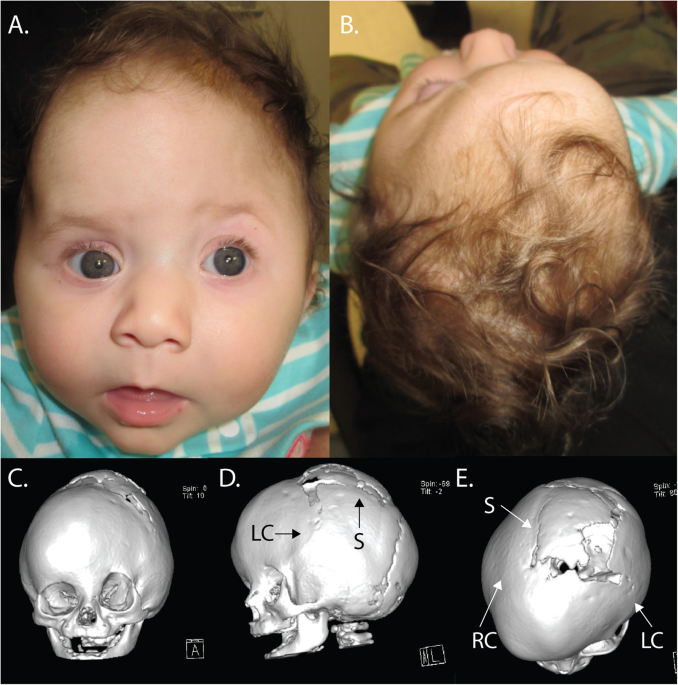
was delivered at term after an uncomplicated pregnancy, and was referred to the pediatric neurosurgical service at birth due to her abnormal head shape. On physical exam, the patient had
flattening of the left frontal bone with contralateral frontal bossing and associated harlequin deformity of the left orbit, consistent with left coronal synostosis. In addition, the
calvarium posterior to the coronal sutures was elongated and narrow (scaphocephalic), consistent with sagittal synostosis (Fig. 1a, b). A CT scan confirmed sagittal and left coronal
synostosis, and the child underwent endoscopic strip craniectomy at nine weeks of age. The child was subsequently followed biweekly in neurosurgery clinic and by an orthotist for helmet
adjustments to shape skull growth. Two months after surgery, the orthotist noted that the child’s left forehead remained flattened and was not rounding as expected. At the child’s subsequent
neurosurgical clinic visit, the parietal and occipital regions had rounded and expanded as expected; however, the left frontal region had stopped rounding. A repeat head CT was performed,
which demonstrated rapid healing and patency of the sagittal suture and re-fusion of the left coronal suture along with complete fusion of the right coronal suture (Fig. 1c–e). Such rapid
recurrence of craniosynostosis is extremely unusual. To correct the anterior deformity, the child underwent a cranial vault reconstruction with fronto-orbital advancement at six months of
age. The child is developing normally to date, and it is unknown at present if she will need further surgical cranial reconstruction. To explore potential genetic contributions to her
condition, we performed whole exome sequencing of the case-parent trio using DNA prepared from buccal swab samples according to standard protocols. Exome capture was performed using the IDT
xGen capture reagent, which was followed by 99 base paired-end sequencing on the Illumina HiSeq 2000 instrument. Sequence reads were aligned to the GRCh37/hg19 human reference genome using
BWA-Mem. Local realignment and quality score recalibration were performed using the GATK pipeline, after which variants were called using the GATK Haplotype Caller. A Bayesian algorithm,
TrioDeNovo, was used to call de novo mutations1. VQSR ‘PASS’ variants with an ExAC allele frequency ≤10−3 sequenced to a depth of eight or greater in the proband and 10 or greater in each
parent with Phred-scaled genotype likelihood scores >30 and de novo quality scores (log10(Bayes factor)) >6 were considered. Independent aligned reads at variant positions were
visualized in silico to remove false calls. All retained calls had de novo genotype quality scores of 100. Transmitted variants were called as per above, and all variants were annotated
using ANNOVAR2 with allele frequencies assigned to each variant from the ExAC database3. Analysis showed that the proband had rare heterozygous LOF mutations in both of the two predominant
non-syndromic CS genes, _SMAD6_ and _TCF12_4,5. The mutation in _SMAD6_ was an early frameshift mutation (p. 152 fs*27), which was transmitted from an unaffected parent (Fig. 2, Table 1).
The mutation in _TCF12_ was also a frameshift mutation (p.K548fs*14), which was de novo. Both mutations were absent from the ExAC and GnomAD databases, which contain >240,000 alleles3,
and both mutations were confirmed by Sanger sequencing (Fig. 2). No other compelling heterozygous rare LOF or damaging missense variants were identified, and no rare recessive genotypes were
identified (Table 1, Supplementary Table 1). Heterozygous _TCF12_ mutations have been previously shown to cause coronal CS with considerable phenotypic overlap with Saethre–Chotzen
syndrome4, which is caused by LOF mutation in _TWIST1_, which heterodimerizes with _TCF12_ to inhibit transcription downstream of BMP signaling. Similar LOF mutations in _TCF12_ were
subsequently identified in patients with non-syndromic coronal craniosynostosis6. De novo or transmitted LOF mutations in _SMAD6_ are found in ~6% of non-syndromic midline craniosynostosis
cases5. LOF mutations in both _TCF12_ and _SMAD6_ both show striking incomplete penetrance (~40 and 20% penetrance, respectively)4,7. In the case of _SMAD6_, epistatic interaction with a
common risk variant near BMP2, which by itself has modest effect on risk, increases penetrance to >90%5,7. The _BMP2_ rs1884302 locus was genotyped in the proband and both parents, and no
family members harbored the CS risk allele ‘C’, consistent with the parent harboring the _SMAD6_ mutation being free of CS (Fig. 2). The combination of rare LOF mutations at established
Mendelian loci (_SMAD6_ and _TCF12_) in the proband was particularly interesting8. While _SMAD6_ has long been known as an inhibitory-SMAD that negatively regulates BMP signaling, _TCF12_
silencing in mesenchymal stem cells was only recently shown to result in increased phosphorylation of receptor-SMADs, implying that loss of TCF12 function also augments BMP signaling via the
BMP/SMAD axis9. This finding suggests that the combination of _SMAD6_ and _TCF12_ haploinsufficiency increases BMP signaling to levels substantially greater than those seen with either
mutation alone, sufficient to ensure penetrance at both the sagittal and coronal sutures (Fig. 3). Moreover, while neither _SMAD6_ nor _TCF12_ haploinsufficiency in isolation has been
associated with increased rates of reoperation4,5, we propose that these mutations together promote sufficiently high osteogenic drive to promote the very unusual rapidity of recurrent
synostosis after surgery (Figs. 1, 3). The combination of a common _BMP2_ variant with LOF variants in SMAD6 is sufficient to push BMP/SMAD signaling to levels sufficient to cause suture
fusion5. The present results suggest that alleles other than the common _BMP2_ risk variant can have epistasis with rare _SMAD6_ alleles. It seems compelling that the combination of a
_SMAD6_ LOF mutation with loss of an independent inhibitor of BMP signaling via _TCF12_ mutation produces particularly high BMP/SMAD signaling and a strikingly more severe phenotype than
_SMAD6_ LOF mutation alone (Figs. 1, 3). It will be interesting to see whether other patients with non-syndromic complex CS also have mutations in these two genes. While several factors,
such as patient age at presentation, suture fusion pattern, and patient co-morbidities, play a role in which type of surgery (endoscopic versus open) is offered to craniosynostosis patients,
knowing the genetic results for specific patients could prove useful in guiding operative planning for this complex patient population. The goals of cranial vault reconstruction are to
obtain an aesthetically pleasing shape of the skull that will allow adequate growth of the brain in ideally one operation. Identification of high risk genotypes prior to surgery—particularly
in cases with unusual clinical features—may prove useful in guiding surgical management in the future and may enable more informed discussions with patients’ families. HGV DATABASE The
relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2330 https://doi.org/10.6084/m9.figshare.hgv.2333. REFERENCES
* Wei, Q. et al. A Bayesian framework for de novo mutation calling in parents-offspring trios. _Bioinformatics_ 31, 1375–1381 (2015). Article CAS Google Scholar * Wang, K., Li, M. &
Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. _Nucleic Acids Res._ 38, e164 (2010). Article Google Scholar * Lek, M. et al.
Analysis of protein-coding genetic variation in 60,706 humans. _Nature_ 536, 285–291 (2016). Article CAS Google Scholar * Sharma, V. P. et al. Mutations in TCF12, encoding a basic
helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. _Nat. Genet._ 45, 304–307 (2013). Article CAS Google Scholar * Timberlake, A. T. et al. Two locus
inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. _ELife_ 5, e20125 (2016). Article Google Scholar * Wilkie, A. O. M., Johnson, D. & Wall,
S. A. Clinical genetics of craniosynostosis. _Curr. Opin. Pediatr._ 29, 622–628 (2017). Article CAS Google Scholar * Timberlake, A. T. et al. De novo mutations in inhibitors of Wnt, BMP,
and Ras/ERK signaling pathways in non-syndromic midline craniosynostosis. _Proc. Natl Acad. Sci. USA_ 114, E7341–E7347 (2017). Article CAS Google Scholar * Posey, J. E. et al. Resolution
of disease phenotypes resulting from multilocus genomic variation. _N. Engl. J. Med._ 376, 21–31 (2017). Article CAS Google Scholar * Yi, S., Yu, M., Yang, S., Miron, R. J. & Zhang,
Y. Tcf12, a member of basic helix-loop-helix transcription factors, mediates bone marrow mesenchymal stem cell osteogenic differentiation in vitro and in vivo. _Stem Cells_ 35, 386–397
(2017). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS The study protocol was approved by the Yale Human Investigation Committee Institutional Review Board, and consent
for use of patient photographs was obtained by the treating physicians. This project was supported by the Yale Center for Mendelian Genomics (NIH Grant M#UM1HG006504-05), the NIH Medical
Scientist Training Program (NIH/National Institute of General Medical Sciences Grant T32GM007205), and the Howard Hughes Medical Institute. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS *
Department of Genetics, Yale University School of Medicine, New Haven, CT, USA Andrew T. Timberlake, Carol Nelson-Williams, Charuta G. Furey & Richard P. Lifton * Section of Plastic and
Reconstructive Surgery, Yale University School of Medicine, New Haven, CT, USA Andrew T. Timberlake, Robin Wu & John A. Persing * Division of Pediatric Neurosurgery, University of North
Carolina School of Medicine, Chapel Hill, NC, USA Kristi I. Hildebrand & Scott W. Elton * Division of Plastic and Reconstructive Surgery, University of North Carolina School of Medicine,
Chapel Hill, NC, USA Jeyhan S. Wood * Laboratory of Human Genetics and Genomics, The Rockefeller University, New York, NY, USA Richard P. Lifton Authors * Andrew T. Timberlake View author
publications You can also search for this author inPubMed Google Scholar * Robin Wu View author publications You can also search for this author inPubMed Google Scholar * Carol
Nelson-Williams View author publications You can also search for this author inPubMed Google Scholar * Charuta G. Furey View author publications You can also search for this author inPubMed
Google Scholar * Kristi I. Hildebrand View author publications You can also search for this author inPubMed Google Scholar * Scott W. Elton View author publications You can also search for
this author inPubMed Google Scholar * Jeyhan S. Wood View author publications You can also search for this author inPubMed Google Scholar * John A. Persing View author publications You can
also search for this author inPubMed Google Scholar * Richard P. Lifton View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR
Correspondence to Richard P. Lifton. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer
Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. ELECTRONIC SUPPLEMENTARY MATERIAL SUPPLEMENTARY TABLE 1 RIGHTS AND PERMISSIONS
OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or
other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Timberlake, A.T., Wu, R.,
Nelson-Williams, C. _et al._ Co-occurrence of frameshift mutations in _SMAD6_ and _TCF12_ in a child with complex craniosynostosis. _Hum Genome Var_ 5, 14 (2018).
https://doi.org/10.1038/s41439-018-0014-x Download citation * Received: 25 April 2018 * Revised: 29 May 2018 * Accepted: 02 June 2018 * Published: 28 June 2018 * DOI:
https://doi.org/10.1038/s41439-018-0014-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative