Ot-82, a novel anticancer drug candidate that targets the strong dependence of hematological malignancies on nad biosynthesis
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Effective treatment of some types of cancer can be achieved by modulating cell lineage-specific rather than tumor-specific targets. We conducted a systematic search for novel agents
selectively toxic to cells of hematopoietic origin. Chemical library screenings followed by hit-to-lead optimization identified OT-82, a small molecule with strong efficacy against
hematopoietic malignancies including acute myeloblastic and lymphoblastic adult and pediatric leukemias, erythroleukemia, multiple myeloma, and Burkitt’s lymphoma in vitro and in mouse
xenograft models. OT-82 was also more toxic towards patients-derived leukemic cells versus healthy bone marrow-derived hematopoietic precursors. OT-82 was shown to induce cell death by
inhibiting nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in the salvage pathway of NAD synthesis. In mice, optimization of OT-82 dosing and dietary niacin further
expanded the compound’s therapeutic index. In toxicological studies conducted in mice and nonhuman primates, OT-82 showed no cardiac, neurological or retinal toxicities observed with other
NAMPT inhibitors and had no effect on mouse aging or longevity. Hematopoietic and lymphoid organs were identified as the primary targets for dose limiting toxicity of OT-82 in both species.
These results reveal strong dependence of neoplastic cells of hematopoietic origin on NAMPT and introduce OT-82 as a promising candidate for the treatment of hematological malignancies.
SIMILAR CONTENT BEING VIEWED BY OTHERS FROM A DRUG REPOSITIONING TO A STRUCTURE-BASED DRUG DESIGN APPROACH TO TACKLE ACUTE LYMPHOBLASTIC LEUKEMIA Article Open access 29 May 2023
ANTI-LEUKEMIA EFFECTS OF OMIPALISIB IN ACUTE MYELOID LEUKEMIA: INHIBITION OF PI3K/AKT/MTOR SIGNALING AND SUPPRESSION OF MITOCHONDRIAL BIOGENESIS Article 11 October 2023 INHIBITION OF
NICOTINAMIDE PHOSPHORIBOSYLTRANSFERASE (NAMPT) WITH OT-82 INDUCES DNA DAMAGE, CELL DEATH, AND SUPPRESSION OF TUMOR GROWTH IN PRECLINICAL MODELS OF EWING SARCOMA Article Open access 10
September 2020 INTRODUCTION In the United States alone, nearly 60,000 people die from hematopoietic (HP) malignancies such as leukemia, lymphoma, and myeloma annually [1]. Despite some
advancements, treatments for many HP malignancies remain insufficiently efficacious and associated with high toxicity. In addition to acute toxicities, genotoxic treatments have long-term
negative effects on health and quality of life, including an increased risk for development of secondary cancers [2]. Therefore, new therapies with improved efficacy and safety are urgently
needed. Some effective anticancer agents are directed against tissue-specific targets rather than cancer-specific targets. For example, _l_-asparaginase- and CD20-targeting drugs [3, 4] are
widely used in hematological oncology and are considered “anti-tissue/lineage” agents since they do not distinguish between normal and transformed cells and act against lineage-specific
metabolic deficiency or lineage-specific surface antigen, respectively. To identify new potential drugs of this type, we screened chemical libraries using a cell-based phenotypic assay for
HP tissue-specific cytotoxic agents. A small molecule identified in this screen was found to be an inhibitor of the ubiquitous metabolic enzyme nicotinamide phosphoribosyltransferase
(NAMPT). NAMPT is the rate-limiting enzyme in the salvage pathway by which nicotinamide adenine dinucleotide (NAD) is synthesized from nicotinamide. NAD can also be synthesized de novo from
tryptophan or via an alternative salvage pathway from nicotinic acid (NA) or NA riboside. Since the NAMPT-dependent salvage pathway is the major pathway used in mammalian cells, NAMPT
inhibition results in the depletion of NAD, which is essential for energy metabolism, oxidation–reduction reactions, and signaling pathways that regulate gene expression, DNA repair, and
calcium homeostasis [5,6,7]. In general, NAMPT is overexpressed in cancer cells compared with corresponding normal cells, presumably reflecting their heightened metabolic and signaling
requirements. In addition, higher NAMPT expression is associated with tumor aggressiveness and poor prognosis [8]. Thus, NAMPT has been defined as an attractive target for anticancer
therapy. Multiple previously identified NAMPT inhibitors have shown efficacy in cancer models, including ovarian, colorectal, HP, prostatic, pancreatic and non-small cell lung carcinomas,
neuroblastoma and fibrosarcoma [9,10,11,12,13,14,15] (reviewed in [16]). The NAMPT inhibitors FK866, GMX1778 (CHS828) and GMX1778’s prodrug, GMX1777, were evaluated in clinical trials in
patients with advanced solid tumors, but these did not go beyond Phase II due to serious toxicities, including thrombocytopenia [17, 18]. Nevertheless, targeting of NAMPT remains an
attractive strategy that is being explored by multiple drug development teams [16]. Here we describe OT-82, a new NAMPT inhibitor with marked efficacy against HP malignancies, a favorable
pharmacological profile and a high therapeutic index. METHODS CELL CULTURE AND REAGENTS Cell lines were obtained from ATCC (Manassas, VA, USA) or Sigma Aldrich (St Louis, MO, USA) and were
periodically tested for the lack of mycoplasma contamination. Cells were maintained in RPMI 1640 or DMEM with phenol red supplemented with 10% fetal bovine serum, 100 units/mL penicillin,
100 μg/mL streptomycin, and 2 mM l-glutamine in a humidified atmosphere containing 5% CO2 at 37 °C. Normal human bone marrow mononuclear cells (BM-MNC) were obtained from ALLCELLS, LLC
(Almeda CA, USA). Patients’ BM-MNC were procured at the Hematologic Bank of RPCCC and NAMPT monoclonal antibody (clone OMNI 379) was obtained from Cayman Chemicals (Ann Arbor, MI, USA).
FK866 was purchased from Selleckchem (Houston, TX, USA). OT-82 has been custom synthesized by Nanosyn Inc (Santa Rosa, CA, USA). HIGH-THROUGHPUT SCREENING High-throughput screening was
performed using >200,000 compounds from libraries of synthetic small molecules from Chembridge Corporation (San Diego, CA, USA) and Maybridge Corporation (Altrincham, UK) as described in
Supplementary Methods. CYTOTOXICITY ASSAY AND CHARACTERIZATION OF CELL DEATH The effect of compounds on cell viability was assessed by resazurin assay (see Supplementary Methods). Caspase-3
activity was determined using the Caspase Fluorometric (AMC) Substrate/Inhibitor QuantiPak BML-AK005 kit (Enzo Life Sciences, Farmingdale, NY, USA) following the manufacturer’s instructions.
Two-Step Cell Cycle analysis and Mitochondrial Membrane Potential Assay were performed using a NC-3000 cytometer following the manufacturer’s instructions (Chemometec, Gydevang, Denmark;
Application note 3001) as described in Supplementary Methods. IDENTIFICATION AND VALIDATION OF THE MOLECULAR TARGET OF OT-82 NAMPT was identified as the protein target of OT-82 by affinity
chromatography followed by mass spectroscopy (see Supplementary Methods). NAMPT activity was measured using the Cyclex NAMPT Colorimetric Assay Kit (MBL International, Woburn, MA, USA). NAD
was measured using Enzyfluo NAD/NADH assay kit (BioAssays Systems, Hayward, CA, USA). ATP was measured using ATPlite Luminescence ATP detection kit (Perkin Elmer, Waltham, MA, USA). All
assays were performed following the kit manufacturer’s instructions. ANIMAL STUDIES Efficacy studies in mouse models were performed according to protocols approved by the Roswell Park Cancer
Institute (RPCI) Institutional Animal Care and Use Committee (IACUC) and the Animal Care and Ethics Committee of the University of New South Wales. Toxicity studies performed in mice and
nonhuman primates (Macaca fascicularis) were conducted by Pharmaron (Beijing, China) in accordance with Pharmaron’s IACUC policies and procedures. Studies in mouse subcutaneous and systemic
xenograft models are described in the Supplementary Methods. Physiological Frailty Index (PFI) was measured in mice as previously described [19]. STATISTICAL ANALYSIS Data are reported as
mean values ± standard error. For animal studies, mean tumor volume and mean body (or organ) weights were compared between groups using two-sided unpaired Student’s _t_-test (GraphPad
Prism5). In patient-derived xenograft models, event-free survival (EFS) curves were compared between groups by Gehan–Wilcoxon test (R statistical software). _p_ values ≤0.05 were considered
significant. RESULTS ISOLATION AND OPTIMIZATION OF SMALL MOLECULES WITH SPECIFIC TOXICITY TOWARDS HEMATOPOIETIC CANCER CELLS To identify the compounds with selective toxicity against HP
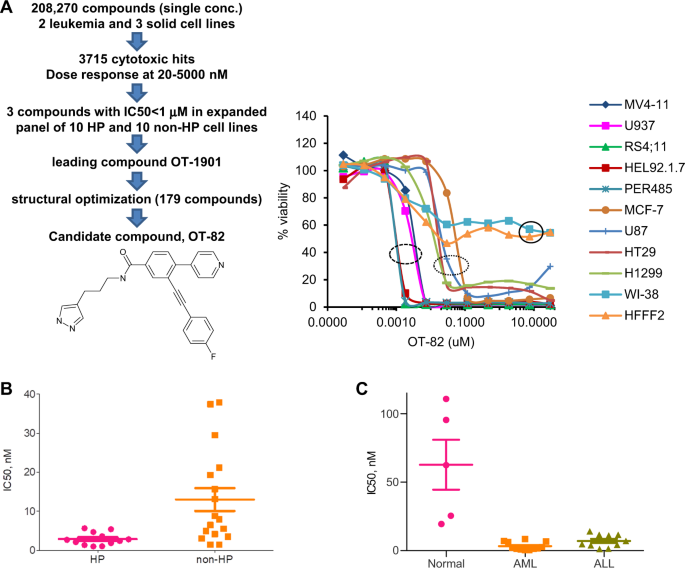
cancer cells, we performed a cell-based high-throughput screening of more than 200,000 small molecules (Fig. 1a and Supplementary Methods). The three most active compounds (with IC50 values
<1000 nM) were further characterized using a panel of 12 HP and 17 non-HP cell lines. Compound OT-1901 showed the best combination of activity (IC50 = 26.3 ± 5.6 nM) and selectivity (on
average, 7.3-fold less toxic to non-HP vs HP cells). A series of 179 proprietary structural analogs of OT-1901 were synthesized and tested for activity and selectivity against HP cancer
cells, as well as improved physico-chemical properties. From these, we selected OT-82 (Fig. 1a) as our lead drug candidate. The spectrum of OT-82 cytotoxicity was determined in vitro using
12 HP cancer and 17 non-HP (breast, prostate, pancreatic, cervical, ovarian, colon and lung carcinomas, melanoma and glioblastoma) human cancer-derived cell lines (Table 1, Fig. 1a, b). The
average IC50 for OT-82 was significantly higher in non-HP cancer cells compared with HP cancer cells (13.03 ± 2.94 nM vs 2.89 ± 0.47 nM; _p_ < 0.008) (Table 1; Fig. 1b). While OT-82 was
cytotoxic towards all types of neoplastic cells tested, the drug had a cytostatic effect on normal diploid fibroblasts (WI-38 and HFFF2 strains), even at concentrations as high as 30 μM
(Fig. 1a). In addition, BM-MNC from healthy donors were significantly less sensitive to OT-82 than BM-MNC from leukemia patients (IC50 = 62.69 ± 18.20 nM for healthy donors vs 3.31 ± 0.85 nM
for AML and 7.10 ± 1.47 nM for ALL, _p_ < 0.001 for both; Fig. 1c). Thus, in vitro, OT-82 demonstrated tissue-selective (HP vs non-HP) as well as cancer-selective (tumor vs normal)
cytotoxicity. EFFICACY OF OT-82 IN PRECLINICAL MODELS OF HEMATOLOGIC MALIGNANCIES Next, we tested the therapeutic effect of OT-82 in vivo, using subcutaneous (SC) and systemic mouse
xenograft models of HP malignancies. Oral (PO) administration of OT-82 (25 or 50 mg/kg) for 6 consecutive days per week for 3 weeks caused dose-dependent suppression of SC growth of human
AML-derived MV4–11 cells (Fig. 2a). The 25 mg/kg dose led to a threefold reduction in mean tumor volume by day 17 of treatment (385 mm2 vs. 1135 mm2, _p_ = 0.0001), while the higher dose
resulted in complete eradication of tumors within 10 days of treatment initiation with no relapses during 43 days after the discontinuation of treatment. Tumors that grew in the 25 mg/kg
OT-82-treated group grew more slowly than those in the vehicle-treated group and remained sensitive to higher doses of the compound, undergoing near complete regression by day 17 after mice
were switched to the 50 mg/kg dose (Fig. 2a). A similar OT-82 regimen was also effective in a SC xenograft model of human erythroleukemia (HEL92.1.7, Fig. 2b), with 50 mg/kg dosing reducing
mean tumor volume from 961 mm2 (control) to 219 mm2 (_p_ = 0.0003) by the end of treatment. OT-82 was also efficacious in systemic leukemia models generated by intravenous (IV) infusion of
MV4-11 and HEL92.1.7 cells, with PO administration 6 days/week for 10 weeks significantly prolonging mouse survival (Fig. 2c). In the AML model (MV4-11), 25 and 40 mg/kg OT-82 dosing
increased median survival by 83 days (161%) and 87 days (168%), respectively (_P_ < 0.0001 vs control for both), with 30 and 40% of mice remaining leukemia-free (vs 0% in vehicle-treated
group). For the erythroleukemia model (HEL92.1.7), median survival following 40 mg/kg OT-82 treatment was extended by 80 days (233%; _P_ < 0.0001). OT-82 was also highly effective in a
patient-derived xenograft model of high-risk pediatric ALL [20,21,22,23,24]. Mice systemically engrafted (IV infusion) with cells from an infant MLL-rearranged ALL patient were treated with
50 mg/kg OT-82 PO 3 days/week for 6 weeks. OT-82 significantly prolonged EFS of treated mice with a median leukemia growth delay (LGD = median EFStreated minus median EFScontrol) of more
than 57 days (_p_ < 0.0001; Fig. 2c). According to clinical response criteria (Supplementary Table S1), OT-82 induced a complete response (Supplementary Table S2). To optimize OT-82
treatment, we varied the duration of consecutive administration and the interval lengths between treatments. Since in vitro studies showed that at least 24–48 h of constant incubation with
the compound are necessary to detect >90% in NAD and ATP depletion (see below), we used 2 days as the minimal period of consecutive treatment. We compared the following regimens applied
as three cycles of days of treatment (t)/days of rest (r): 3t/4r; 2t/5r; 3t/7r; and 2t/8r in SCID mice bearing MV4–11 subcutaneous xenografts (Fig. 3a). All four regimens resulted in the
elimination of tumors after three treatment cycles. The 3t/4r regimen was identified as optimal based on the proportion of mice experiencing relapse within 60 days after treatment
discontinuation (5% and 20% in 80 and 60 mg/kg treatment groups, respectively, vs higher proportions for all other regimens). Increased efficacy of OT-82 applied using the optimized 3t/4r
regimen was confirmed in the systemic model of AML (MV4-11). With this regimen, 100% and 56% of mice treated for 3 weeks with 40 or 20 mg/kg OT-82 survived 90 days after treatment
discontinuation, respectively (Fig. 3b), as compared with 40% survival in groups given 40 mg/kg OT-82 6 days per week (6t/1r) for 10 weeks (Fig. 2c). Notably, improved survival was observed
with the optimized regimen even though the total drug exposure was dramatically reduced (360 mg/kg for 3 weeks of 3t/4r 40 mg/kg dosing vs. 2,400 mg/kg for 10 weeks of 6t/1r 40 mg/kg
dosing). The optimized OT-82 3t/4r regimen also potently inhibited tumor growth in SC xenograft models of Burkitt’s lymphoma (Fig. 3c) and multiple myeloma (Fig. 3d) and was highly effective
in a systemic PDX model of pediatric Philadelphia chromosome-positive (Ph+) ALL (Fig. 3e) [20, 22]. In the latter model, OT-82 (40 mg/kg, 3t/4r) was more effective (achieved Maintained
Complete Response, Supplementary Tables 1 and 2) than a 3-drug standard induction therapy for ALL (vincristine/dexamethasone/_l_-asparaginase; VXL), which failed to induce a therapeutic
response (Objective Response Measure = Progressive Disease 2). Neither of the above-described regimens caused detectable weight loss in treated animals as evident from the graphs presented
in Supplementary Fig. S1. MOLECULAR TARGET AND MECHANISM OF CYTOTOXICITY OF OT-82 The molecular target of OT-82 was identified as NAMPT by affinity chromatography of MV4-11 cellular extracts
exposed to immobilized active OT-82 and an inactive structural analog followed by mass spectrometry and western blotting with anti-NAMPT antibody (Fig. 4a). Functional NAMPT inhibition by
OT-82 was confirmed in an in vitro assay using recombinant human NAMPT (Fig. 4a). This showed the IC50 of OT-82 to be similar to that of the well-described NAMPT inhibitor FK866 (41 and 39
nM, respectively) [11]. Further confirmation of NAMPT as the target of OT-82 was provided by utilization of the alternative salvage pathway to synthesize NAD from NA to protect against the
toxic effects of NAMPT inhibition: both leukemia cell lines tested were completely protected from OT-82-induced cell death (up to 10 mM OT-82) by addition of 10 μM NA to the culture medium
(Fig. 4a). Consistent with the role of NAMPT as a rate-limiting enzyme in the major NAD biosynthesis pathway and the requirement of NAD for ATP production, OT-82 treatment of MV4-11 cells
caused dose-dependent reductions in cellular NAD and ATP concentrations (with expected delay) that were consistent with OT-82’s IC50 for cell viability (Fig. 4b, Table 1). In examining the
mechanism of OT-82-induced cell death, we found that in vitro treatment of MV4-11 cells with OT-82 for 48 h resulted in activation of caspase-3, an increase in the proportion of cells with
sub-G1 DNA content, and depolarization of the mitochondrial membrane, all hallmarks of apoptotic cell death (Fig. 4b). The OT-82 concentration producing a 50% relative effect was essentially
the same in these three assays and was similar to the IC50 for cell viability. Together, these observations indicate that OT-82 induces apoptotic cell death via NAMPT inhibition leading to
NAD and ATP depletion. EFFECT OF DIETARY NIACIN ON THE THERAPEUTIC INDEX OF OT-82 Niacin in the form of NA is the substrate for an alternative (NAMPT-independent) salvage pathway for NAD
biosynthesis. Given the redundancy of NAD synthesis pathways and the ability of NA to protect leukemia cells from OT-82-induced cell death in vitro (Fig. 4d), we hypothesized that reduced
dietary niacin might increase OT-82 efficacy in vivo. To test this, we evaluated OT-82 efficacy in mice fed custom purified Teklad diets (Harlan) containing different percentages (100, 45,
or 15%) of the recommended daily amount of niacin (30 mg/kg [25]) or the regular diet used at RPCI (2018S, Harlan), which contains 115 mg/kg niacin (380% of the recommended level)
(Supplementary Table S3). To determine the ED50 of OT-82 for each diet, SCID mice bearing SC MV4-11 xenografts were treated PO with vehicle or OT-82 (2–40 mg/kg, 3t/4r for 3 weeks) and tumor
growth was monitored as described in the Supplementary Methods. In parallel, the 50% lethal dose (LD50) was determined by monitoring survival of nontumor bearing SCID mice in the four diet
groups treated with OT-82 (25–200 mg/kg, 3t/4r for 3 weeks). Both the ED50 (indicating efficacy) and the LD50 (indicating toxicity) of OT-82 decreased as dietary niacin was reduced. However,
diet-dependent reductions in ED50 were greater than those in LD50, resulting in an overall increase in therapeutic index (calculated as LD50/ED50). Consistent with this, 82% of mice on the
regular Harlan diet treated with 40 mg/kg OT-82 relapsed with tumors within 60 days post treatment, while none or only 17% of mice in reduced-niacin diet groups relapsed (Supplementary Table
S3). The inverse relationship between dietary niacin (NA, vitamin B3) and anticancer efficacy of OT-82 shown here supports adherence to diets with no more than 100% of the recommended daily
amount of niacin for optimal OT-82 efficacy. PHARMACOLOGICAL AND TOXICOLOGICAL PROFILE OF OT-82 To advance OT-82 towards clinical development, we assessed pharmacological and toxicological
characteristics of this drug candidate in mice and nonhuman primates (NHPs, Cynomolgus Monkeys) selected as having similar to human metabolic profiles of OT-82 (see Supplementary Methods and
Supplementary Table S4). Recombinant mouse and human NAMPTs in biochemical assays in vitro showed similar their sensitivity to the inhibitory action of OT-82 (Supplementary Fig. S2). Cells
derived from both species selected for the toxicological studies showed roughly similar sensitivity to NAMPT inhibitors (Supplementary Fig. S3). The pharmacokinetics and biodistribution of
OT-82 after oral administration were evaluated using LC/MS-MS. The maximum plasma concentrations of OT-82 in mice (dose 10 mg/kg) and NHP (50 mg/kg) were 525 and 1049 ng/ml respectively and
were observed at 1 and 3.33 h respectively with a half-life (_T_1/2) of about 1.75 h for both species (Supplementary Table 4). Biodistribution analysis in mice indicated broad
bioavailability of OT-82 with substantial accumulation of OT-82 in all major organs except the brain presumably indicative of inability of the drug to penetrate the blood brain barrier
(Supplementary Fig. S4). Toxicological characteristics of OT-82 in rodents and NHPs were obtained according to Good Laboratory Practice requirements using the projected clinical regimen of
administration. Overall, these studies demonstrated good tolerability of OT-82. At a dose 55 mg/kg, which corresponds to the therapeutic dose in multiple mouse models used, no toxicity of
the drug has been documented. At the highest doses used (90 mg/kg for mice and 30 mg/kg for NHP) damage in HP and lymphoid organs (bone marrow, spleen, and lymph nodes) with signs of
hepatotoxicity were registered in both species. All toxicological effects were reversible, and the measured parameters returned to normal levels within 2 weeks after treatment completion
(Supplementary Tables S5 and S6; Supplementary Fig. S5). Since potential retinal and cardiac toxicities were reported for NAMPT inhibitors GNE-617 and GMX1778 [16, 26, 27] and NAMPT-mediated
NAD+ biosynthesis was found to be essential for vision in mice [28], we compared ophthalmological and cardiological effects in C57BL/6 mice of physiologically similar sublethal doses of
OT-82 (100 mg/kg) or GMX1778 (125 mg/kg) given PO for 5 consecutive days. While both compounds caused similar body and spleen weight decreases (Fig. 5a, b), only GMX1778-treated animals
demonstrated morphological signs of retinal toxicity (reduced thickness of the outer nuclear and outer plexiform layers of the retina, roughness of the external limiting membrane, and
numerous pale spaces in the rods and cones layer indicative of cell loss). Consistently, no indications of retinal damage were observed by in-life ophthalmological examination or
histopathological assessment of eye tissues in experiments with chronic administration of OT-82 in mice and NHPs. Potential cardiac toxicity of OT-82 in comparison with GMX1778 was evaluated
by histological analysis of H&E-stained sections of heart tissue collected 3 h after the last drug treatment. Compared with vehicle-treated controls, GMX1778-treated mice showed
degenerative changes to the myocardium indicated by local myofibrils degeneration. In contrast, hearts from OT-82-treated mice did not show any type of morphological damage. In NHP studies
using implanted telemetry, there were no signs of cardiac toxicity at 10 or 30 mg/kg OT-82 daily (3 days), with 60 mg/kg/day resulting in transient QT prolongation, registered between 2 and
6 h post the third dosing, with ECG parameters reversed to normal by 8 h. This dose exceeds more than 2 times the dose equivalent planned as the maximal dose for a prospective human phase I
study. Thus, unlike previously studied NAMPT inhibitors, OT-82 does not cause retinal or cardiac toxicity within a clinically relevant range of doses. EFFECT OF OT-82 TREATMENT ON FRAILTY
INDEX AND LONGEVITY OF MICE NAD levels have been shown to decline in aging tissues and this decline has been linked to age-related pathologies stemming from malfunctioning of NAD-dependent
factors involved in cellular homeostasis (e.g., sirtuins and PARP) [29,30,31]. Moreover, pharmacological elevation of NAD by increasing dietary niacin was reported to reverse aging
biomarkers in mice [32]. This raises a concern that NAMPT inhibitors might accelerate aging and increase the risk of age-related diseases. To address this, we assessed the effect of OT-82
treatment on mouse longevity (natural lifespan) and biological age. Objective quantitative determination of the biological age of animals is possible through a recently developed cumulative
health assessment parameter, PFI [19]. As evident from Fig. 6, treatment of aged (87 weeks old) C57BL/6 male and female mice with five courses of OT-82 mimicking the established optimal
regimen did not affect longevity or PFI, thus providing additional support for the overall safety of OT-82. DISCUSSION Molecular targets for effective anticancer treatment are typically
regulators of proliferation (e.g., RAS or MYC family members, growth factor receptors, etc.) or cell viability (e.g., BCL2 family members, NF-κB pathway regulators, etc.) that are aberrantly
expressed in tumors due to structural mutations, amplifications or other types of deregulation and are essential for tumor cell growth or viability [33,34,35]. Antagonists of such factors
can distinguish between normal and transformed cells and thus may have potential as anticancer drugs. However, there are also examples of effective anticancer drugs that do not distinguish
between normal and cancer cells, but rather act against tissue-specific targets. Obviously, such drugs can only be directed against tumors originating from tissues that are either not
essential for organism viability (e.g., gender-specific tissues) or can be regenerated from intrinsic or transplanted progenitors. The clinical success of this class of “anti-tissue” drugs
reflects the failure of tumors to deviate from their epigenetic origin, remaining dependent on tissue-specific factors. Antagonists of androgen and estrogen receptors (e.g., enzalutamide
[36, 37] and tamoxifen [38], respectively) used for treatment of prostate and breast cancers, respectively, belong to this category. Tissue-specific drugs are broadly used in hematologic
oncology. These include agents targeting lineage-specific antigens (e.g., anti-CD20 monoclonal antibody rituximab [4] and CD19-targeting CAR T-cell therapies tisagenlecleucel and
axicabtagene [39]) or exploiting lineage-specific metabolic deficiencies (e.g., _l_-asparaginase, which blocks supply of the essential amino acid asparagine to myeloid cells deficient in its
production [3, 40]). Genotoxic preconditioning by total body irradiation, melphalan, or busulfan (conventionally used in preparation for bone marrow transplantation) is another
tissue-specific form of treatment for HP malignancies, capitalizing on the sensitivity of HP cells to DNA damage-mediated apoptosis [41]. NAMPT has been extensively studied as an anticancer
target, but without specific focus on HP malignancies [16] except for a recently published demonstration of high sensitivity of MLL-translocated leukemias to NAMPT inhibition [9], an
observation that is consistent with our data. NAMPT is not unique in being a tissue-specific target with ubiquitous expression. For example, androgen receptor is expressed in multiple
tissues, but its blockade is essentially cytotoxic only for prostate epithelial cells [42]. Hence, tissue-specificity is determined by a target’s function rather than its expression.
However, there is an important distinction between NAMPT and other tissue-specific targets: in addition to being more critical for the viability of HP versus other cell types, NAMPT
inhibition is also more toxic towards malignant versus normal cells of the same HP origin. Thus, NAMPT uniquely combines properties of both tissue specificity and cancer specificity.
Sensitivity of cells to OT-82 did not depend on their proliferation rate (Supplementary Fig. S6) indicating that other mechanisms determine selective toxicity of NAMPT inhibition to cancer
cells and particularly HP malignancies presumably involving cell type-specific metabolic dependency. Being generally less energy efficient than normal cells [43], tumor cells are expected to
be more sensitive to ATP depletion, which unavoidably follows NAMPT suppression. However, OT-82-mediated cytotoxicity showed dependence on the NAD rather than ATP concentration (Fig. 4b)
favoring alternative explanations. For example, NAD deficiency affects the function of sirtuins, a class of histone deacetylases [44, 45], and PARP family members that also use NAD as a
co-factor [46, 47]. Thus, in addition to causing energy depletion, NAMPT inhibitors combine the activities of two classes of proven anticancer agents—histone deacetylases and PARP inhibitors
[45, 47]. Greatly increased sensitivity to NAMPT inhibitors was observed in tumor cells with IDH1/2 mutations known to be associated with reduced baseline levels of NAD) compared with
IDH1/2-wild type cells [48]. Thus, mutation of IDH1/2 may represent a predictor of tumor sensitivity to NAMPT inhibitors (IDH1 and IDH2 mutations have been reported in 10–33% of AMLs,
>80% of gliomas, >50% chondrosarcomas, and >10% cholangiocarcinomas [49,50,51]). IDH1/2-mutant AML is an especially attractive target due to the higher dependency of HP malignant
cells on NAD status. While increased sensitivity of cells with IDH1/2 mutations to NAMPT inhibitors has a plausible explanation, the higher susceptibility of HP cells, and particularly
transformed HP cells, to NAMPT depletion remains to be explained. Despite several attempts, no NAMPT inhibitors have been approved as anticancer drugs [18]. Dose limiting toxicities revealed
in phase I trials predominantly involved the HP system, which is consistent with the tissue-specific elevated NAMPT dependence described in this study. Clinical trials of previously
identified NAMPT inhibitors were mostly conducted in patients with solid tumors [18]. The hematological complications observed in these trials would likely have been more easily handled in
patients with HP malignancies in whom these symptoms are common and expected. Furthermore, the treatment schedules used in most published trials are substantially different from the optimal
regimen determined in our work (e.g., APO866 was administered by 96 h intravenous infusion every 4 weeks [17]; CHS828 was administered orally 1×/week for 4 weeks [18]). By focusing on HP
malignancies and optimized regimens, it is highly possible that NAMPT inhibitors could show sufficient safety and efficacy to advance in clinical development. Additional opportunities to
improve the efficacy of this class of agents include modulation of diet and selection of patients based on tumor IDH1/2 status. Finally, OT-82 represents a structurally new class of NAMPT
inhibitors that may avoid some of the pitfalls of its predecessors, as illustrated by the reduced retinal toxicity of OT-82 observed in our experiments, which is likely to reflect lesser
ability of OT-82 to penetrate blood–retinal barrier [52] rather than differences in its mechanisms of action from other NAMPT inhibitors. In fact, A2780 cells selected for the resistance to
OT-82 were equally resistant to FK866 and vice versa (Supplementary Fig. S7) suggesting similarity in molecular mechanisms of NAMPT inhibition by these otherwise structurally very different
compounds. Overall, our work provides renewed support for the feasibility of pharmacological NAMPT inhibition as an anticancer treatment approach by: (i) describing a new inhibitor with a
superior set of properties; (ii) directing clinical development of NAMPT inhibitors specifically towards HP malignancies; (iii) suggesting an effective treatment regimen; and (iv) reducing
fears of possible systemic toxicity of NAMPT inhibitors stemming from data showing the importance of NAD levels in aging [53, 54]. In particular, our results indicate the strong clinical
potential of OT-82 for safe and effective treatment of hematological malignancies further highlighted in the accompanying manuscript [55]. REFERENCES * Siegel RL, Miller KD, Jemal A. Cancer
statistics, 2017. CA Cancer J Clin. 2017;67:7–30. PubMed Google Scholar * Aronson JK. Meyler's Side Effects of Drugs, 16th edn, Elsevier Science, Oxford, UK, 2015. * Asselin B,
Rizzari C. Asparaginase pharmacokinetics and implications of therapeutic drug monitoring. Leuk Lymphoma. 2015;56:2273–80. CAS PubMed PubMed Central Google Scholar * Van Meerten T,
Hagenbeek A. CD20-targeted therapy: a breakthrough in the treatment of non-Hodgkin’s lymphoma. Neth J Med. 2009;67:251–9. p. PubMed Google Scholar * Galli U, Travelli C, Massarotti A,
Fakhfouri G, Rahimian R, Tron GC, et al. Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J Med Chem. 2013;56:6279–96. CAS PubMed Google Scholar *
Chiarugi A, Dölle C, Felici R, Ziegler M. The NAD metabolome—a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–52. p. CAS PubMed Google Scholar * Bogan KL, Brenner C.
Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD + precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115–30. CAS PubMed Google Scholar *
Shackelford RE, Mayhall K, Maxwell NM, Kandil E, Coppola D. Nicotinamide phosphoribosyltransferase in malignancy: a review. Genes Cancer. 2013;4:447–56. PubMed PubMed Central Google
Scholar * Matheny CJ, Wei MC, Bassik MC, Donnelly AJ, Kampmann M, Iwasaki M, et al. Next-generation NAMPT inhibitors identified by sequential high-throughput phenotypic chemical and
functional genomic screens. Chem Biol. 2013;20:1352–63. CAS PubMed Google Scholar * Watson M, Roulston A, Belec L, Billot X, Marcellus R, Bedard D, et al. The small molecule GMX1778 is a
potent inhibitor of NAD + biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol Cell Biol. 2009;29:5872–88. CAS PubMed PubMed
Central Google Scholar * Nahimana A, Attinger A, Aubry D, Greaney P, Ireson C, Thougaard AV, et al. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic
malignancies. Blood. 2009;113:3276–86. CAS PubMed Google Scholar * O’Brien T, Oeh J, Xiao Y, Liang X, Vanderbilt A, Qin A, et al. Supplementation of nicotinic acid with NAMPT inhibitors
results in loss of in vivo efficacy in NAPRT1-deficient tumor models. Neoplasia. 2013;15:1314–29. PubMed PubMed Central Google Scholar * Xiao Y, Elkins K, Durieux JK, Lee L, Oeh J, Yang
LX, et al. Dependence of tumor cell lines and patient-derived tumors on the NAD salvage pathway renders them sensitive to NAMPT inhibition with GNE-618. Neoplasia. 2013;15:1151–60. PubMed
PubMed Central Google Scholar * Fuchs D, Rodriguez A, Eriksson S, Christofferson R, Sundberg C, Azarbayjani F. Metronomic administration of the drug GMX1777, a cellular NAD synthesis
inhibitor, results in neuroblastoma regression and vessel maturation without inducing drug resistance. Int J Cancer. 2010;126:2773–89. CAS PubMed Google Scholar * Chini CCS, Guerrico AMG,
Nin V, Camacho-Pereira J, Escande C, Barbosa MT, et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res.
2014;20:120–30. CAS PubMed Google Scholar * Sampath D, Zabka TS, Misner DL, O’Brien T, Dragovich PS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy
in cancer. Pharm Ther. 2015;151:16–31. CAS Google Scholar * Holen K, Saltz LB, Hollywood E, Burk K, Hanauske AR. The pharmacokinetics, toxicities, and biologic effects of FK866, a
nicotinamide adenine dinucleotide biosynthesis inhibitor. Investig N Drugs. 2008;26:45–51. CAS Google Scholar * von Heideman A, Berglund A, Larsson R, Nygren P. Safety and efficacy of NAD
depleting cancer drugs: results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother Pharm. 2010;65:1165–72. CAS Google Scholar * Antoch MP, Wrobel M,
Kuropatwinski KK, Gitlin I, Leonova KI, Toshkov I, et al. Physiological frailty index (PFI): quantitative in-life estimate of individual biological age in mice. Aging. 2017;9:615–26. CAS
PubMed PubMed Central Google Scholar * Szymanska B, Wilczynska-Kalak U, Kang MH, Liem NLM, Carol H, Boehm I, et al. Pharmacokinetic modeling of an induction regimen for in vivo combined
testing of novel drugs against pediatric acute lymphoblastic leukemia xenografts. PLoS ONE. 2012;7:e33894. CAS PubMed PubMed Central Google Scholar * Suryani S, Bracken LS, Harvey RC,
Sia KCS, Carol H, Chen I-M, et al. Evaluation of the in vitro and in vivo efficacy of the JAK inhibitor AZD1480 against JAK-mutated acute lymphoblastic leukemia. Mol Cancer Ther.
2015;14:364–74. CAS PubMed Google Scholar * Liem NLMM, Papa RA, Milross CG, Schmid MA, Tajbakhsh M, Choi S, et al. Characterization of childhood acute lymphoblastic leukemia xenograft
models for the preclinical evaluation of new therapies. Blood. 2004;103:3905–14. CAS PubMed Google Scholar * Lock RB, Liem N, Farnsworth ML, Milross CG, Xue C, Tajbakhsh M, et al. The
nonobese diabetic/severe combined immunodeficient (NOD/SCID) mouse model of childhood acute lymphoblastic leukemia reveals intrinsic differences in biologic characteristics at diagnosis and
relapse. Blood. 2002;99:4100–8. CAS PubMed Google Scholar * Houghton PJ, Lock R, Carol H, Morton CL, Phelps D, Gorlick R, et al. Initial testing of the hypoxia-activated prodrug PR-104 by
the pediatric preclinical testing program. Pediatr Blood Cancer. 2011;57:443–53. PubMed Google Scholar * Terakata M, Fukuwatari T, Kadota E, Sano M, Kanai M, Nakamura T, et al. The niacin
required for optimum growth can be synthesized from L-tryptophan in growing mice lacking tryptophan-2,3-dioxygenase. J Nutr. 2013;143:1046–51. CAS PubMed Google Scholar * Zabka TS, Singh
J, Dhawan P, Liederer BM, Oeh J, Kauss MA, et al. Retinal toxicity, in vivo and in vitro, associated with inhibition of nicotinamide phosphoribosyltransferase. Toxicol Sci. 2015;144:163–72.
CAS PubMed Google Scholar * Misner DL, Kauss MA, Singh J, Uppal H, Bruening-Wright A, Liederer BM, et al. Cardiotoxicity associated with nicotinamide phosphoribosyltransferase inhibitors
in rodents and in rat and human-derived cells lines. Cardiovasc Toxicol. 2017;17:307–18. CAS PubMed Google Scholar * Lin JB, Kubota S, Ban N, Yoshida M, Santeford A, Sene A, et al.
NAMPT-mediated NAD + biosynthesis is essential for vision in mice. Cell Rep. 2016;17:69–85. CAS PubMed PubMed Central Google Scholar * Verdin E. NAD + in aging, metabolism, and
neurodegeneration. Science. 2015;350:1208–13. CAS PubMed Google Scholar * Verdin E. The many faces of Sirtuins: coupling of NAD metabolism, sirtuins and lifespan. Nat Med. 2014;20:25–7.
CAS PubMed Google Scholar * Li J, Bonkowski MS, Moniot S, Zhang D, Hubbard BP, Ling AJY, et al. A conserved NAD + binding pocket that regulates protein-protein interactions during aging.
Science. 2017;355:1312–7. CAS PubMed PubMed Central Google Scholar * Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, et al. Long-term administration of nicotinamide
mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 2016;24:795–806. CAS PubMed PubMed Central Google Scholar * Torti D, Trusolino L. Oncogene addiction as
a foundational rationale for targeted anti-cancer therapy: promises and perils. EMBO Mol Med. 2011;3:623–36. CAS PubMed PubMed Central Google Scholar * Pagliarini R, Shao W, Sellers WR.
Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015;16:280–96. CAS PubMed PubMed Central Google Scholar * Willis RE. Targeted
cancer therapy: vital oncogenes and a new molecular genetic paradigm for cancer initiation progression and treatment. Int J Mol Sci. 2016;17:1552. p. PubMed Central Google Scholar * Helsen
C, Van den Broeck T, Voet A, Prekovic S, Van Poppel H, Joniau S, et al. Androgen receptor antagonists for prostate cancer therapy. Endocr Relat Cancer. 2014;21:T105–18. CAS PubMed Google
Scholar * Rathkopf D, Scher HI. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J. 2013;19:43–9. CAS PubMed PubMed Central Google Scholar * Martinkovich S,
Shah D, Planey SL, Arnott JA. Selective estrogen receptor modulators: tissue specificity and clinical utility. Clin Interv Aging. 2014;6:1437–52. * Chow VA, Shadman M, Gopal AK. Translating
anti-CD19 CAR T-cell therapy into clinical practice for relapsed/refractory diffuse large B-cell lymphoma. Blood. 2018;132:777–781. * Pieters R, Hunger SP, Boos J, Rizzari C, Silverman L,
Baruchel A, et al. L-asparaginase treatment in acute lymphoblastic leukemia. Cancer. 2011;117:238–49. p. CAS PubMed Google Scholar * Gyurkocza B, Sandmaier BM. Conditioning regimens for
hematopoietic cell transplantation: one size does not fit all. Blood. 2014;124:344–53. CAS PubMed PubMed Central Google Scholar * Inoue S, Mizushima T, Miyamoto H. Role of the androgen
receptor in urothelial cancer. Mol Cell Endocrinol. 2018;465:73–81. CAS PubMed Google Scholar * Epstein T, Gatenby RA, Brown JS. The Warburg effect as an adaptation of cancer cells to
rapid fluctuations in energy demand. PLoS ONE. 2017;12:e0185085. * Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. 2015;15:608–24. * Hu J, Jing
H, Lin H. Sirtuin inhibitors as anticancer agents. Future Med Chem. 2014;6:945–66. CAS PubMed Google Scholar * Morales J, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, et al. Review of poly
(ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr. 2014;24:15–28. CAS PubMed PubMed Central Google
Scholar * Underhill C, Toulmonde M, Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Ann Oncol. 2011;22:268–79. * Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F,
Lelic N, et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell. 2015;28:773–84. CAS PubMed PubMed Central Google Scholar * Molenaar RJ, Maciejewski JP,
Wilmink JW, van Noorden CJF. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene. 2018;37:1949–60. CAS PubMed PubMed Central Google Scholar * Clark O, Yen K, Mellinghoff
IK. Molecular pathways: isocitrate dehydrogenase mutations in cancer. Clin Cancer Res. 2016;22:1837–42. CAS PubMed PubMed Central Google Scholar * Marcucci G, Maharry K, Wu YZ,
Radmacher MD, Mrózek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia
group B study. J Clin Oncol. 2010;28:2348–55. CAS PubMed PubMed Central Google Scholar * Hosoya K, Tomi M, Tachikawa M. Strategies for therapy of retinal diseases using systemic drug
delivery: relevance of transporters at the blood-retinal barrier. Expert Opin Drug Deliv. 2011;8:1571–87. CAS PubMed Google Scholar * Chini CCS, Tarragó MG, Chini EN. NAD and the aging
process: role in life, death and everything in between. Mol Cell Endocrinol. 2017;455:62–74. CAS PubMed Google Scholar * Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends
Cell Biol. 2014;24:464–71. CAS PubMed PubMed Central Google Scholar * Somers K, Evans K, Cheung L, Karsa M, Pritchard T, Kosciolek A, et al. Effective targeting of NAMPT in
patient-derived xenograft models of high-risk pediatric acute lymphoblastic leukemia. Leukemia. (2019). https://doi.org/10.1038/s41375-019-0683-6. Download references ACKNOWLEDGEMENTS This
manuscript is dedicated to the memory of Dr Meir Wetzler. We are grateful to Eunice Wang for providing samples from the Hematologic Procurement Bank of Roswell Park Comprehensive Cancer
Center. We thank Patricia Stanhope Baker for help in manuscript preparation. This work was supported by grants from Cancer Australia APP1164865 to MH, MJH, KS, and MN; from Cancer Council
NSW PG16-01 to MJH, MH, MN, and RL; from the National Cancer Institute CA199222 and CA199000, The National Health and Medical Research Council of Australia NHMRC Fellowships APP1059804 and
APP1157871, Cancer Australia and Kids’ Cancer Project APP1164865, Tenix Foundation and the ISG Foundation to RL and from Anthony Rothe Memorial Trust to RL and MJH; from the National Cancer
Institute grant P30CA016056 involving the use of Clinical Data Network and Laboratory Animals Shared Resources to Roswell Park Cancer Center. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS *
Oncotartis, Inc., Buffalo, NY, USA Lioubov Korotchkina, Denis Kazyulkin, Pavel G. Komarov, Alex Polinsky, Ekaterina L. Andrianova, Sangeeta Joshi, Mahima Gupta, Slavoljub Vujcic, Eugene
Kononov, Ilia Toshkov, Yuan Tian, Peter Krasnov & Olga B. Chernova * Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA Mikhail V. Chernov, Jean Veith, Marina P. Antoch &
Andrei V. Gudkov * Children’s Cancer Institute, Sydney, NSW, Australia Shiloh Middlemiss, Klaartje Somers, Richard B. Lock, Murray D. Norris, Michelle J. Henderson & Michelle Haber *
University of New South Wales Centre for Childhood Cancer Research, Sydney, NSW, Australia Murray D. Norris Authors * Lioubov Korotchkina View author publications You can also search for
this author inPubMed Google Scholar * Denis Kazyulkin View author publications You can also search for this author inPubMed Google Scholar * Pavel G. Komarov View author publications You can
also search for this author inPubMed Google Scholar * Alex Polinsky View author publications You can also search for this author inPubMed Google Scholar * Ekaterina L. Andrianova View
author publications You can also search for this author inPubMed Google Scholar * Sangeeta Joshi View author publications You can also search for this author inPubMed Google Scholar * Mahima
Gupta View author publications You can also search for this author inPubMed Google Scholar * Slavoljub Vujcic View author publications You can also search for this author inPubMed Google
Scholar * Eugene Kononov View author publications You can also search for this author inPubMed Google Scholar * Ilia Toshkov View author publications You can also search for this author
inPubMed Google Scholar * Yuan Tian View author publications You can also search for this author inPubMed Google Scholar * Peter Krasnov View author publications You can also search for this
author inPubMed Google Scholar * Mikhail V. Chernov View author publications You can also search for this author inPubMed Google Scholar * Jean Veith View author publications You can also
search for this author inPubMed Google Scholar * Marina P. Antoch View author publications You can also search for this author inPubMed Google Scholar * Shiloh Middlemiss View author
publications You can also search for this author inPubMed Google Scholar * Klaartje Somers View author publications You can also search for this author inPubMed Google Scholar * Richard B.
Lock View author publications You can also search for this author inPubMed Google Scholar * Murray D. Norris View author publications You can also search for this author inPubMed Google
Scholar * Michelle J. Henderson View author publications You can also search for this author inPubMed Google Scholar * Michelle Haber View author publications You can also search for this
author inPubMed Google Scholar * Olga B. Chernova View author publications You can also search for this author inPubMed Google Scholar * Andrei V. Gudkov View author publications You can
also search for this author inPubMed Google Scholar CONTRIBUTIONS AVG, OBC, AP, ELA, DK, RBL, MDN, MJH and MH designed the study. LK, DK, PGK, ELA, SJ, MG, SV, EK, IT, YT, PK, MVC, JV, MPA,
SM and KS participated in study conduct and data collection. LK, DK, PGK, AP, ELA, IT, PK, MVC, MPA, KS, RBL, MDN, MJH, MH, OBC and AVG analyzed and interpreted the data. LK, ELA, KS, RBL,
MDN, MJH, MH, OBC and AVG wrote and/or made critical revisions to the manuscript. CORRESPONDING AUTHOR Correspondence to Andrei V. Gudkov. ETHICS DECLARATIONS CONFLICT OF INTEREST This work
was sponsored by Oncotartis, Inc. (Buffalo, NY, USA). Oncotartis representatives were involved in study design and conduct; data collection, management, analysis, and interpretation;
manuscript preparation, review, and approval; and the decision to submit the manuscript for publication. All authors had access to primary study data. LK, DK, PGK, AP, ELA, SJ, MG, SV, EK,
IT, YT, PK, MPA, OBC, and AVG were employees or consultants of Oncotartis and received compensation in the form of salary, consulting fees, stock options, or sponsored research. ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL
MATERIAL RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if
changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the
material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to
obtain permission directly from the copyright holder. To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE
THIS ARTICLE Korotchkina, L., Kazyulkin, D., Komarov, P.G. _et al._ OT-82, a novel anticancer drug candidate that targets the strong dependence of hematological malignancies on NAD
biosynthesis. _Leukemia_ 34, 1828–1839 (2020). https://doi.org/10.1038/s41375-019-0692-5 Download citation * Received: 30 May 2019 * Revised: 23 November 2019 * Accepted: 06 December 2019 *
Published: 02 January 2020 * Issue Date: July 2020 * DOI: https://doi.org/10.1038/s41375-019-0692-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative