Suppression of tak1 pathway by shear stress counteracts the inflammatory endothelial cell phenotype induced by oxidative stress and tgf-β1
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Endothelial dysfunction is characterised by aberrant redox signalling and an inflammatory phenotype. Shear stress antagonises endothelial dysfunction by increasing nitric oxide
formation, activating anti-inflammatory pathways and suppressing inflammatory pathways. The TAK1 (MAP3K7) is a key mediator of inflammation and non-canonical TGF-β signalling. While the
individual roles of TAK1, ERK5 (MAPK7) and TGF-β pathways in endothelial cell regulation are well characterised, an integrative understanding of the orchestration of these pathways and their
crosstalk with the redox system under shear stress is lacking. We hypothesised that shear stress counteracts the inflammatory effects of oxidative stress and TGF-β1 on endothelial cells by
restoring redox balance and repressing the TAK1 pathway. Using human umbilical vein endothelial cells, we here show that TGF-β1 aggravates oxidative stress-mediated inflammatory activation
and that shear stress activates ERK5 signalling while attenuating TGF-β signalling. ERK5 activation restores redox balance, but fails to repress the inflammatory effect of TGF-β1 which is
suppressed upon TAK1 inhibition. In conclusion, shear stress counteracts endothelial dysfunction by suppressing the pro-inflammatory non-canonical TGF-β pathway and by activating the ERK5
pathway which restores redox signalling. We propose that a pharmacological compound that abates TGF-β signalling and enhances ERK5 signalling may be useful to counteract endothelial
dysfunction. SIMILAR CONTENT BEING VIEWED BY OTHERS Β-CATENIN PROMOTES ENDOTHELIAL SURVIVAL BY REGULATING ENOS ACTIVITY AND FLOW-DEPENDENT ANTI-APOPTOTIC GENE EXPRESSION Article Open access
30 June 2020 THE MITOCHONDRIAL CA2+ UNIPORTER CHANNEL SYNERGIZES WITH FLUID SHEAR STRESS TO INDUCE MITOCHONDRIAL CA2+ OSCILLATIONS Article Open access 07 December 2022 ENDOTHELIAL
Γ-PROTOCADHERINS INHIBIT KLF2 AND KLF4 TO PROMOTE ATHEROSCLEROSIS Article Open access 04 September 2024 INTRODUCTION The vascular endothelium is a monolayer of cells that acts as the
regulatory interface between blood and the vessel wall. Given the capability to receive and respond to biochemical as well as biomechanical stimuli, the endothelium is a key regulator of
cardiovascular homeostasis1. Adverse alterations of the endothelial phenotype (endothelial dysfunction) precede the pathogenesis of cardiovascular disorders, particularly atherosclerosis1,2
and pulmonary hypertension3. The maintenance of a healthy endothelial phenotype relies on a delicate balance between nitric oxide (NO) production and reactive oxygen species (ROS) formation,
both of which are crucial to the maintenance of cellular redox tone and the functioning of redox-related cell signalling. A decreased NO bioavailability, secondary to enhanced NO
degradation by ROS can tip the redox balance and cause impaired NO-mediated signalling, an early hallmark of endothelial dysfunction2,4. In physiology, the phenotype of endothelial cells is
tightly regulated by their responses to mechanical forces, especially shear stress5,6,7. Shear stress exerted by laminar blood flow increases NO bioavailability, while reducing ROS
production. Therefore, shear stress safeguards endothelial redox homeostasis and counteracts endothelial dysfunction5,7,8. The protective effects of shear stress on endothelial cells extend
to its inhibition of inflammatory signalling cascades, such as nuclear factor kappa-light-chain-enhancer of activated B cell (NFκB)5,9 and p38 mitogen-activated protein kinase (MAPK)10
pathways. The expression of inflammatory entities, such as adhesion molecules and chemoattractants, that are activated by these signalling pathways, is also inhibited by shear
stress5,6,7,8,9. Shear stress also elicits its protective effects through activation of mitogen-activated protein kinase 7 (MAPK7), also known as extracellular signal-regulated kinase 5
(ERK5)7. ERK5 signalling downregulates inflammatory entities through induction of the anti-inflammatory transcription factors, Kruppel-like factor 2 (KLF2)11 or KLF412. Notably, TGF-β
signalling also mediates shear-induced KLF2 expression through the activin receptor-like kinase 5 (ALK5)/SMAD pathway13,14. While the individual roles of NFkB, p38 MAPK, ERK5 and TGF-β
pathways in endothelial dysfunction are well delineated, an understanding of the orchestration of these pathways and their crosstalk with the redox system in the context of relevant
haemodynamic forces remain obscure. In addition to activating the canonical SMAD pathway, TGF-β also activates the non-canonical mitogen-activated protein kinase kinase kinase 7 (MAP3K7),
also known as TGF-β-activated kinase 1 (TAK1) pathway15. Activation of TAK1 by inflammatory cytokines induces the expression of inflammatory entities in endothelial cells9. Surprisingly, the
consequences of TAK1 activation for endothelial cells upon TGF-β stimulation and its regulation by shear stress remain unknown. The levels of oxidative stress and TGF-β1 increase upon
vascular damage4,16. However, the molecular mechanisms by which shear stress regulates the phenotype of endothelial cells upon oxidative stress and TGF-β1 stimulation are poorly understood.
Our earlier studies revealed that shear stress suppresses a severe form of endothelial dysfunction, TGF-β-induced endothelial-to-mesenchymal transition (EndMT), through ERK5 activation17.
Here, we hypothesised that shear stress counteracts the inflammatory effects of oxidative stress and TGF-β1 on endothelial cells by repressing the TAK1 pathway and by restoring redox
balance. To test this hypothesis, we subjected human umbilical vein endothelial cells (HUVEC) to the pro-inflammatory (ROS) and pro-fibrotic (TGF-β1) triggers, and dissected the associated
cell signalling responses of ERK5, ALK5 and TAK1 to shear stress using a combination of molecular biological, biochemical and pharmacological tools. RESULTS TGF-Β1 AGGRAVATES THE
INFLAMMATORY EFFECTS OF OXIDATIVE STRESS Studies about the combined effects of bovine brain extract (referred to as endothelial cell growth factors, ECGF throughout the text) deprivation and
TGF-β1 on redox balance and inflammation are scarce. Therefore, we investigated the influence of ECGF deprivation and TGF-β1 stimulation on ROS and NO metabolites formation, as well as the
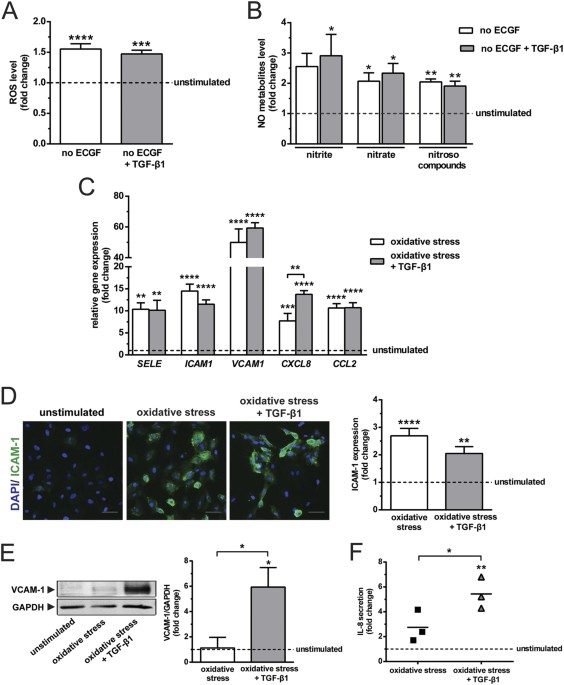
subsequent expression of inflammatory molecules by HUVEC. ECGF deprivation caused a 1.6-fold increase of intracellular ROS formation, but TGF-β1 stimulation did not further affect ROS
induction (Fig. 1A). Interestingly, ECGF deprivation also increased NO production as evidenced by an elevation of nitrite, nitrate and nitroso compounds, whereas TGF-β1 had no added effect
(Fig. 1B). Consistent with previous report18, these results demonstrate that increases in oxidative stress are counterbalanced by an up-regulation of endogenous NO production, and that
TGF-β1 has insignificant influence on this redox reaction. The oxidative stress caused by ECGF deprivation was associated with an increase in the expression of inflammatory molecules, _SELE_
(10.3-fold), _ICAM1_ (14.5-fold), _VCAM1_ (50-fold), _CXCL8_ (7.7-fold) and _CCL2_ (10.7-fold; Fig. 1C). Of note, TGF-β1 caused an additional increase of _CXCL8_ expression (Fig. 1C).
Oxidative stress alone or in conjunction with TGF-β1 stimulation did not alter the expression of _TNFA_ and _IL1B_. However, the combined stimulation with oxidative stress and TGF-β1
increased _IL6_ expression by 1.7-fold (see Supplementary Fig. S1A). Oxidative stress induced the protein expression of ICAM-1 (2.7-fold), but there was no added effect of TGF-β1 on this
induction (Fig. 1D). Of note, oxidative stress alone did not alter the expression VCAM-1, while oxidative stress together with TGF-β1 caused a 6-fold upregulation (Fig. 1E). TGF-β1
synergised with oxidative stress in inducing IL-8 secretion, as shown by the 2-fold higher induction of IL-8 secretion upon treatment with TGF-β1 compared to oxidative stress alone (Fig.
1F). These data indicate that dependent on the redox status of endothelial cells, the effects of TGF-β1 on inflammatory molecules expression are variable. Endothelial cells stimulated by
oxidative stress and TGF-β1 had 27-fold higher interaction with leukocytes, as compared with the unstimulated control (see Supplementary Fig. S1B). Of note, this inflammatory endothelial
phenotype was endowed with the feature of EndMT, as shown by the upregulation of mesenchymal markers, _ACTA2, TAGLN_ and _CNN1,_ as well as the downregulation of _PECAM1, THBD_ and _NOS3_
(see Supplementary Fig. S1C). LAMINAR SHEAR STRESS SUPPRESSES THE INFLAMMATORY EFFECTS OF OXIDATIVE STRESS AND TGF-Β1 To assess the regulation of endothelial phenotype by shear stress, we
exposed endothelial cells to laminar shear stress at a magnitude of 20 dynes/cm2. Shear stress enhanced endothelial NO production (Fig. 2A), down-regulated the expression of _SELE_
(3.3-fold)_, VCAM1_ (1.4-fold)_, CXCL8_ (19.2-fold) and _CCL2_ (23.8-fold), while up-regulated the expression of _ICAM1_ (2.3-fold; Fig. 2B). In agreement with the gene expression data,
shear stress downregulated VCAM-1 protein expression (Fig. 2C). IL-8 secretion did not change in response to shear stress (Fig. 2D). Upon oxidative stress and additional TGF-β1 stimulation,
sheared endothelial cells had a 6-, 11.5-, 94- and 42-fold downregulation of _SELE, VCAM1, CXCL8_ and _CCL2_ (Fig. 2E), respectively, when compared with the static control. Notably, shear
stress repressed the upregulation of VCAM-1 (Fig. 2F) and IL-8 (Fig. 2G) protein expression by 12.7- and 5.3-fold, respectively. These data demonstrate that shear stress attenuates the
combined inflammatory effects of oxidative stress and TGF-β1, and this effect is likely mediated via an increase in NO production. ACTIVATION OF ERK5 REDUCES OXIDATIVE STRESS, BUT DOES NOT
REPRESS THE INFLAMMATORY EFFECTS OF TGF-Β1 We were intrigued to elucidate as to how ERK5 signalling influences the combined effects of oxidative stress and TGF-β1, and vice versa in terms of
endothelial phenotype regulation. To address this, we examined cellular redox state and phenotype of MEK5D-transduced cells subjected to oxidative stress and stimulated with TGF-β1. MEK5D
is a constitutively active mutant of MEK5 that induces sustained activation of the ERK5 pathway12. Sustained activation of ERK5 suppressed the generation of both, ROS (Fig. 3A) and NO
production (Fig. 3B). TGF-β1 did not alter the effects of ERK5 on maintenance of redox poise. Activation of ERK5 inhibited the expression of _SELE, CXCL8_ and _CCL2_, but up-regulated the
expression of _ICAM1_ and _VCAM1_ (Fig. 3C). Upon treatment with TGF-β1, expression of _ICAM1_ and _VCAM1_ in MEK5D-transduced cells were enhanced by 2- and 39-fold, respectively (Fig. 3C).
Notably, TGF-β1 synergised with the ERK5-mediated upregulation of _ICAM1_ and _VCAM1_. In spite of the enhanced transcript expression, TGF-β1 had negligible effect on protein expression of
ICAM-1 in MEK5D-transduced cells (Fig. 3D). Activation of ERK5 induced the expression of VCAM-1 by a factor of 5 (Fig. 3E). Remarkably, upregulation of VCAM-1 was further enhanced (3.5-fold)
when ERK5 pathway was activated upon TGF-β1 stimulation (Fig. 3E). The TGF-β-induced IL-8 secretion was strongly inhibited (14.6-fold) when MEK5D was stably expressed (Fig. 3F).
Collectively, our data indicate that despite the marked modulatory effects of ERK5 pathway on the magnitude of combined ROS and NO production, it only partially rescues the TGF-β1-induced
alterations in endothelial phenotype. SHEAR STRESS ANTAGONISES THE ACTIVATION OF CANONICAL TGF-Β SIGNALLING, INDEPENDENT OF ERK5 Under stimulation with TGF-β1, shear stress repressed,
whereas ERK5 signalling augmented the expression of VCAM-1. These differences prompted us to dissect the underlying mechanisms. In spite of the TGF-β1 stimulation, both sheared and
MEK5D-transduced cells showed increased ERK5 phosphorylation, as well as enhanced KLF2 and KLF4 expression (Fig. 4A). Others reported that KLF2 attenuates canonical TGF-β signalling through
reduction of SMAD2 phosphorylation and inhibition of SMAD3/4 transcriptional activity19. We therefore investigated whether shear stress downregulates canonical TGF-β signalling while induces
KLF2 expression via ERK5 activation. Our data show that TGF-β1 stimulation induced SMAD2 phosphorylation, an effect that was repressed approximately 1.5-fold by shear (Fig. 4B). This
repression was not caused by the reduced expression of total SMAD2 (Fig. 4B). Activation of ERK5 signalling under static conditions did not suppress the phosphorylation of SMAD2 (Fig. 4C).
Upon stimulation with TGF-β1, MEK5D-transduced cells showed a 2.3-fold higher phosphorylation of SMAD2 than the vector controls (Fig. 4C), implying that the TGF-β signalling was reinforced
by activation of the ERK5 pathway, the mechanism of which is unclear. Sustained ERK5 activation did not alter the expression of total SMAD2 (Fig. 4C). Furthermore, there was a 4-fold
increased expression of SMAD2 target gene, _TAGLN_ in MEK5D-transduced cells under stimulation with TGF-β1 (see Supplementary Fig. S2). These results demonstrate that shear stress
suppresses, whereas ERK5 signalling augments the activation of canonical TGF-β signalling. Shear stress induced the expression of the TGF-β canonical inhibitors, _SMAD6_ and _SMAD7_ 2.4- and
4.1-fold (Fig. 4D), respectively. Shear-induced _SMAD6_ and _SMAD7_ expression was not altered by TGF-β1 stimulation (Fig. 4D). In concordance with shear stress, constitutive activation of
ERK5 (MEK5D-transduced cells) under static condition enhanced the expression of _SMAD6_ and _SMAD7_ 2.4- and 4.5-fold, respectively. Intriguingly, TGF-β1 stimulation augmented the
upregulation of _SMAD6_ and _SMAD7_ in MEK5D-transduced cells (Fig. 4E), supporting the notion that ERK5 signalling enhances the activation of TGF-β signalling. These data show that both
shear stress and activation of ERK5 induce the expression of inhibitory SMADs (I-SMADs), _i.e. SMAD6_ and _SMAD7_. INHIBITION OF CANONICAL TGF-Β SIGNALLING SUPPRESSES OXIDATIVE STRESS, BUT
DOES NOT INFLUENCE THE EXPRESSION OF ICAM-1, VCAM-1 AND IL-8 Since shear stress represses the activation of TGF-β signalling, we inhibited the canonical ALK5/SMAD pathway with the ALK5
inhibitor, SB431542, to investigate whether canonical TGF-β signalling affects redox balance and endothelial phenotype. Inhibition of ALK5/SMAD signalling reduced ROS formation (1.3-fold;
Fig. 5A), but had insignificant effect on NO production (Fig. 5B). ALK5/SMAD signalling had no effect on the expression of _SELE_ and _VCAM1_ (Fig. 5C). Notably, _ICAM1, CXCL8_ and _CCL2_
were almost 2-fold downregulated (Fig. 5C). Inhibition of the ALK5/SMAD signalling had no effects on the induced expression of ICAM-1 (Fig. 5D), VCAM-1 (Fig. 5E) and IL-8 (Fig. 5F) upon
TGF-β1 stimulation. Similarly, the expression of ICAM-1 (Fig. 5G) and VCAM-1 (Fig. 5H) in MEK5D-transduced cells was unaffected by the inhibition of ALK5. These data implicate a role for
ALK5/SMAD signalling in ROS generation, but not in NO production and protein expression of inflammatory molecules. INHIBITION OF TAK1 PATHWAY OR MITOCHONDRIAL ROS PRODUCTION SUPPRESSES THE
UPREGULATION OF ICAM-1, VCAM-1 AND IL-8 Because the inhibition of canonical ALK5/SMAD signalling did not suppress the induced expression of ICAM-1, VCAM-1 and IL-8, we hypothesised that
shear stress inhibits the expression of these molecules by regulating either the non-canonical TAK1 pathway or redox poise. The TAK1 inhibitor, 5z-7-oxozeaenol and the mitochondrial ROS
inhibitor, YCG063, both reduced the formation of intracellular ROS by 1.2- and 1.4- fold, respectively (Fig. 6A). Inhibition of TAK1 and mitochondrial ROS reduced nitrate generation by 2.2-
and 1.3-fold, respectively, but had insignificant effects on nitrite formation (Fig. 6B). Of note, the production of nitrosated species increased dramatically (more than 30-fold) as a result
of mitochondrial ROS inhibition, in the presence (see Supplementary Fig. S3) and absence (Fig. 6B) of constitutive ERK5 activation; these effects were not accompanied by prominent enhanced
nitrite/nitrate formation, indicating they were not caused by increased NO production. While both pathway of formation and functional significance of this finding remain unclear at present,
this data are testimony to marked alterations in mitochondrial nitrosative stress triggered by YCG063. 5Z-7-oxozeaenol suppressed the induced expression of _SELE, ICAM1, VCAM1, CXCL8_ and
_CCL2_ by 5.8-, 4.5-, 15.3-, 20.5-, 2.6-fold, respectively (Fig. 6C). Similarly, YCG063 also downregulated the induced expression of _SELE, ICAM1, VCAM1, CXCL8, CCL2_ by 2.6-, 3.9-, 33.9-,
12.7-, 3.5-fold, respectively (Fig. 6C). Treatment with 5z-7-oxozeaenol or YCG063, downregulated the induction of ICAM-1 by 3-fold (Fig. 6D). The increased level of VCAM-1 was downregulated
by 14.6- and 113.2-fold in response to the treatment with 5z-7-oxozeaenol and YCG063, respectively (Fig. 6E). Inhibition of TAK1 or mitochondrial ROS formation resulted in a 1.9- and
4.3-fold reduction in the level of IL-8 secretion (Fig. 6F). Under the treatment with 5z-7-oxozeaenol or YCG063, the ICAM-1 expression of MEK5D-transduced cells were repressed almost 2-fold
(Fig. 6G). VCAM-1 expression of MEK5D-transduced cells was downregulated by 3.4- and 6.1-fold in response to 5z-7-oxozeaenol or YCG063 treatment, respectively (Fig. 6H). Taken together,
these data show that TAK1 or generation of mitochondrial ROS, or both contribute to oxidative stress and cause upregulation of inflammatory molecules. Interestingly, 5z-7-oxozeaenol or
YCG063 repressed the upregulation of smooth muscle 22α (SM22α) (see Supplementary Fig. S4), suggesting that the TAK1 pathway and ROS contribute to induction of EndMT20,21 while exerting
inflammatory effects on endothelial cells. INHIBITION OF THE P38 MAPK OR NFΚB PATHWAY SUPPRESSES INFLAMMATORY ENDOTHELIAL PHENOTYPE Finally, we dissected the signalling cascades that act
downstream of TAK1 and their attenuation of oxidative stress and suppression of endothelial inflammation. Endothelial cells subjected to oxidative stress and stimulated with TGF-β1, were
treated with the respective inhibitor of p38 MAPK, SB202190, and of IKKβ, SC514. Inhibition of p38 MAPK decreased the induced formation of intracellular ROS by 1.3-fold. Inhibition of IKKβ
had no effect on ROS formation (Fig. 7A). Inhibition of p38 MAPK or IKKβ had insignificant effect on NO production (Fig. 7B). Inhibition of p38 MAPK decreased the induced expression of
_SELE, ICAM1, VCAM1, CXCL8_ and _CCL2_ by 5.6-, 10.2-, 18.1-, 8.7- and 7.4-fold (Fig. 7B), respectively. Similarly, IKKβ inhibition also decreased the induced expression of _SELE_
(21.5-fold)_, ICAM1_ (13-fold)_, VCAM1_ (38-fold)_, CXCL8_ (20-fold) and _CCL2_ (26-fold; Fig. 7C). Taken together, these data suggest that TAK1 activation preferentially activates p38 MAPK
or NFkB, or both, upon TGF-β1 stimulation to induce an inflammatory endothelial phenotype. DISCUSSION In this study, we dissected the interplay of different components in the network of
mechanotransduction which suppresses the inflammatory endothelial phenotype. We showed that shear stress-activated ERK5 signalling restores the redox state of endothelial cells by adjusting
NO and ROS production, but fails to antagonise the inflammatory activation by TGF-β1. Notably, high shear stress counteracts TGF-β1-induced inflammation by suppressing the non-canonical
TGF-β pathway via TAK1, in an ERK5-independent manner. This indicates that although shear stress-activated ERK5 signalling restores the redox state of endothelial cells, it fails to
antagonise the inflammatory effects of TGF-β1. Furthermore, we showed that shear stress abates the kinase activity of ALK5. By contrast, in the absence of shear stress, constitutive
activation of ERK5 with MEK5D, augmented ALK5 activity while suppressing both, ROS and NO production. This suggests that suppression of TGF-β signalling by shear stress is independent of the
ERK5 pathway and redox state (Fig. 8). Our study suggests that oxidative stress not only contributes to impaired NO bioactivity by scavenging NO, but that rates of endothelial ROS and NO
production are indeed coupled to maintain proper functioning of redox-related signalling including phosphorylation processes; thus, NO production not only compensatorily increased upon
enhanced oxidative stress, but decreased proportionally upon repression of cellular ROS production. Reaction of ROS with NO lead to generation of pro-oxidants, such as peroxynitrite4,22.
Peroxynitrite and other reactive species can alter endothelial phenotype by disrupting cellular redox signalling, as well as by inducing the activation of inflammatory pathways, such as NFκB
and p38 MAPK4 (Fig. 8). Our study demonstrated that TGF-β1 aggravates the inflammatory effects of oxidative stress via the non-canonical TGF-β pathway. Consistent with others, we showed
that activation of ERK5 repressed the induction of _SELE_23_, CCL2_ and IL-812. Intriguingly, upon TGF-β1 stimulation, ERK5 signalling did not inhibit VCAM-1 gene and protein expression
which decreased when the TAK1 signalling axis was inhibited. AMP-activated protein kinase (AMPK) mediates shear stress-induced ERK5 signalling, while increasing NO bioavailability and
reducing oxidative stress24. This mechanistic evidence and our finding link shear stress and the ERK5 pathway with redox homeostasis. We suggest that shear stress represses oxidative
stress-induced inflammation by restoring the redox state via ERK5 pathway, but the suppression of TGF-β1-induced inflammation depends on inactivation of TAK1 signalling (Fig. 8). A selective
inhibition of either NFκB or p38 MAPK downregulated the expression of inflammatory molecules. This coincides with earlier findings that TAK1 requires NFκB25 and p38 MAPK26 to elicit its
downstream effects on endothelial cells (Fig. 8). Activation of TAK1 depends predominantly on the affinity of TNF receptor-associated factor 6 (TRAF6) to TGF-β receptor I (TβRI), such as
ALK5 and stimulation of TGF-β receptor II (TβRII) by TGF-β27,28. Therefore, we postulate that shear stress suppresses the activation of TAK1 and its downstream pathways by interfering either
with the affinity of TRAF6 to ALK5 or with the binding of TGF-β1 to TβRII (Fig. 8). The inhibitory SMADs (SMAD6 and SMAD7) are negative feedback regulators of the TGF-β signalling29.
Earlier study elucidated KLF2 via upregulation of SMAD7, represses phosphorylation of SMAD219. In our study, shear stress-induced _SMAD6_ and _SMAD7_ expression correlated with the
downregulation of SMAD2 phosphorylation, which suggests that shear stress inhibits TGF-β signalling via inhibitory SMADs. Notably, SMAD2 phosphorylation was not attenuated in our
MEK5D-transduced cells, despite the enhanced expression of KLF2, _SMAD6_ and _SMAD7_. The overexpression of a SMAD2 target gene, _TAGLN_ upon TGF-β stimulation in our study explains that the
transcriptional activity of SMAD2 in MEK5D-transduced cells was conserved. These data suggest that shear stress inactivates TGF-β signalling in an ERK5-independent manner. We postulate that
SMAD6 and SMAD7 proteins in MEK5D-transduced cells may have been degraded due to SUMO(small ubiquitin-like modifier)ylation and ubiquitylation upon TGF-β stimulation30. Alternatively, the
post-transcriptional processes of these inhibitory _SMADs_ may be affected by microRNAs31. Collectively, the present study provides compelling evidence that regulation of the endothelial
phenotype by shear stress involves an intricate crosstalk of multiple signalling axes, rather than an alteration of select individual pathways (Fig. 8). Our previous findings elucidated that
shear stress mediates EndMT through the ERK5 pathway17. Here, we show that parallel to signalling through ERK5, which safeguards redox homeostasis, shear stress antagonises the inflammatory
effects of TGF-β1 by suppressing the TAK1 pathway (Fig. 8), suggesting that shear stress counteracts endothelial dysfunction by activating ERK5 pathway and suppressing TGF-β pathway. We
propose that the protective effects of shear stress on endothelial cells can be substituted by a pharmacological compound that enhances ERK5 signalling and abates TGF-β signalling
concurrently. MATERIALS AND METHODS Extended materials and methods are available in supplementary information. CELL CULTURE, STIMULATION AND INHIBITION HUVEC were obtained from Lonza (Breda,
The Netherlands) and the Endothelial Cell Facility of University Medical Center Groningen, The Netherlands. Cells were maintained in endothelial cell medium, composed of, RPMI 1640 basal
medium (Lonza, Verviers, Belgium), supplemented with 20% heat-inactivated foetal bovine serum (Invitrogen/GIBCO, CA, USA), 50 μg/ml endothelial cell growth factors supplement (bovine brain
extract; homemade), 1% penicillin-streptomycin (Sigma-Aldrich, MA, USA), 2 mM L-glutamine (Lonza) and 5 U/ml heparin (Leo Pharma, Ballerup, Denmark). Cells were used for experiments at
passage 6 and 7. Confluent monolayers of HUVEC were stimulated for 48 h with or without 5 or 10 ng/ml citric acid activated-TGF-β1 (Peprotech, NJ, USA; #100–21 C) in RPMI 1640 basal medium,
supplemented with 20% heat-inactivated foetal bovine serum, 2 mM L-glutamine, 5 U/ml heparin and 1% penicillin-streptomycin. Cells treated with endothelial cell medium were harvested as
unstimulated controls, Cells were treated with pharmacological inhibitors for 48 h to inhibit desired signalling pathways. SHEAR STRESS EXPERIMENTS Confluent monolayer of HUVEC in flow
channel, μ-Slide I0.4 Luer (ibidi, Martinsried, Germany) were exposed to 20 dynes/cm2 shear stress for 48 h in endothelial cell medium or RPMI 1640 basal medium, supplemented with 5 or 10
ng/ml TGF-β1, 20% heat-inactivated foetal bovine serum, 2 mM L-glutamine, 5 U/ml heparin and 1% penicillin-streptomycin. Ibidi Pump System was employed for generation of laminar flow. Shear
stress experiments were performed in an incubator at 37 °C with 5% CO2. Cells treated with the same media without exposure to flow were harvested as static controls. RETROVIRAL TRANSDUCTION
A retroviral construct for stable expression of constitutively active rat MEK5-α1 (MEK5D) was generated as described before12. Retroviral transduction of HUVEC was performed as previously
described17. Expanded transduced cells were seeded for experiments as described for wild type cells. ROS MEASUREMENT Cells were treated with 20 μM of 2′,7′-dichloroflorescein diacetate
(Sigma-Aldrich; D6883) and analysed with FACSCaliburTM flow cytometer (BD Biosciences, NJ, USA) at λmax, 520 nm. Average intensity was obtained by subtracting the mean fluorescent intensity
of unstained samples from stained samples. Data of each experimental condition are presented as fold changes in average intensity relative to their respective experimental controls.
QUANTIFICATION OF NITRITE, NITRATE AND TOTAL NITROSO COMPOUNDS Cellular formation of NO was quantified by the accumulation of nitrite, nitrate and total nitroso compounds in the cell culture
media. The level of nitrite and nitrate in aliquots of the media was determined using a dedicated analysis system (ENO-20, EiCom, Japan), as previously described32. The level of nitroso
compounds, comprising N-nitrosamines and S-nitrosothiols in aliquots of the same media was quantified by gas-phase chemiluminescence, as previously described33. Concentration of nitrite
(μM), nitrate (μM) and nitroso compounds (nM) in each media was normalized to the respective numbers of cell in each experimental condition. Normalised data are presented as fold changes in
concentration relative to experimental controls. RNA ISOLATION AND RT-QPCR RNA was isolated with either TRIzol reagent (Invitrogen Corp, CA, USA) or RNA-Bee (Bio-Connect, The Netherlands)
according to the manufacturer’s protocol. Primer sets (see Supplementary Table S1) were used to detect amplimers of interest. Data of each experimental condition are presented as fold
changes in gene expression relative to their respective experimental controls. IMMUNOFLUORESCENT STAINING Mouse anti-human ICAM-1 antibody (hybridoma supernatant; Hu5/3; kindly provided by
Professor Dr. Michael A. Gimbrone Jr, Harvard Medical School, Boston, MA, USA)34,35 and SM22α antibody (1:200; Abcam, Cambridge, UK; ab14106) were used. Stained slides were analysed using
TissueFaxs® Zeiss AxioObserver Z1 Microscope System (TissueGnostics, Vienna, Austria). Mean fluorescent intensity quantification was performed with TissueQuest fluorescence analysis software
(TissueGnostics). At least 500 cells were counted for each experimental condition. Data of each experimental condition are presented as fold changes in expression relative to their
respective experimental controls. IMMUNOBLOTTING Odyssey Immunoblotting System (Li-COR Biosciences, USA) was employed. Antibodies against human VCAM-1 (1:100; Santa Cruz Biotechnology, USA;
sc-8304), ERK5 (1:500; Upstate Cell Signaling Solutions, USA; #07–039), KLF4 (1:500; Santa Cruz Biotechnology; sc-20691), KLF2 (1:500; Santa Cruz Biotechnology; sc-28675), p-SMAD2
(Ser465/467; 1:200; Cell Signaling Technology, USA; #3108), SMAD2/3 (1:200; Cell Signaling Technology; #3102) and GAPDH (1:2000; Abcam, UK; ab9484) were used for detection of target
proteins. Expression of target proteins was normalised to loading control, GAPDH. Data of each experimental condition are presented as fold changes in expression relative to their respective
experimental controls. SANDWICH ENZYME-LINKED IMMUNOSORBENT ASSAY (ELISA) Culture media of different experimental condition were collected. Concentration of IL-8 in media was quantified
with Human IL-8 ELISA MAXTM Standard Sets (BioLegend Inc, CA, USA) according to the manufacturer’s protocol. Concentration of IL-8 in each media was normalized to their respective numbers of
cell in each experimental condition. Data of each experimental condition are presented as fold changes in IL-8 concentration (pg/ml) relative to their respective experimental controls.
STATISTICAL ANALYSIS All experimental data were obtained from two to seven independent experiments with duplicates or triplicates. All data are presented as means + standard error of the
mean (SEM). Statistical analyses were performed with GraphPad Prism (Version 6.01; GraphPad Software, Inc., USA). For two-group comparisons, two-tailed ratio paired t test was performed. For
multiple group comparisons, one-way analysis of variance (ANOVA) followed by Sidak’s post-test were carried out. For multiple categorical group comparisons, two-way ANOVA followed by
Sidak’s post-test were executed. All statistical analyses were performed at the 95% of confidence interval. Differences between means were considered significant when probabilities (P) were
less than 0.05. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Lee, E. S. _et al_. Suppression of TAK1 pathway by shear stress counteracts the inflammatory endothelial cell phenotype
induced by oxidative stress and TGF-β1. _Sci. Rep._ 7, 42487; doi: 10.1038/srep42487 (2017). PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. REFERENCES * Deanfield, J. E., Halcox, J. P. & Rabelink, T. J. Endothelial function and dysfunction: testing and clinical relevance.
_Circulation_ 115, 1285–1295 (2007). Article PubMed Google Scholar * Cai, H. & Harrison, D. G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. _Circ
Res_ 87, 840–844 (2000). CAS PubMed Google Scholar * Lewis, L. J., Hoak, J. C., Maca, R. D. & Fry, G. L. Replication of human endothelial cells in culture. _Science_ 181, 453–454
(1973). Article ADS CAS PubMed Google Scholar * Pacher, P., Beckman, J. S. & Liaudet, L. Nitric oxide and peroxynitrite in health and disease. _Physiol Rev_ 87, 315–424 (2007).
Article CAS PubMed Google Scholar * Hahn, C. & Schwartz, M. A. Mechanotransduction in vascular physiology and atherogenesis. _Nat Rev Mol Cell Biol_ 10, 53–62 (2009). Article CAS
PubMed PubMed Central Google Scholar * Zhou, J., Li, Y.-S. & Chien, S. Shear stress–initiated signaling and its regulation of endothelial function. _Arterioscler Thromb Vasc Biol_ 34,
2191–2198 (2014). Article CAS PubMed PubMed Central Google Scholar * Berk, B. C. Atheroprotective signaling mechanisms activated by steady laminar flow in endothelial cells.
_Circulation_ 117, 1082–1089 (2008). Article PubMed Google Scholar * Hsieh, H.-J., Liu, C.-A., Huang, B., Tseng, A. & Wang, D. Shear-induced endothelial mechanotransduction: the
interplay between reactive oxygen species (ROS) and nitric oxide (NO) and the pathophysiological implications. _J Biomed Sci_ 21, 3 (2014). Article PubMed PubMed Central Google Scholar *
Warboys, C. M., Amini, N., de Luca, A. & Evans, P. C. The role of blood flow in determining the sites of atherosclerotic plaques. _F1000 Med Rep_ 3, 5 (2011). Article PubMed PubMed
Central Google Scholar * Yamawaki, H., Lehoux, S. & Berk, B. C. Chronic physiological shear stress inhibits tumor necrosis factor–induced proinflammatory responses in rabbit aorta
perfused _ex vivo_ . _Circulation_ 108, 1619–1625 (2003). Article CAS PubMed Google Scholar * Parmar, K. M. et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like
factor 2. _J Clin Invest_ 116, 49–58 (2006). Article CAS PubMed Google Scholar * Ohnesorge, N. et al. Erk5 activation elicits a vasoprotective endothelial phenotype via induction of
Kruppel-like factor 4 (KLF4). _J Biol Chem_ 285, 26199–26210 (2010). Article CAS PubMed PubMed Central Google Scholar * Walshe, T. E., dela Paz, N. G. & D’Amore, P. A. The role of
shear-induced transforming growth factor-β signaling in the endothelium. _Arterioscler Thromb Vasc Biol_ 33, 2608–2617 (2013). Article CAS PubMed PubMed Central Google Scholar *
Egorova, A. D. et al. Tgfβ/Alk5 signaling is required for shear stress induced klf2 expression in embryonic endothelial cells. _Dev Dyn_ 240, 1670–1680 (2011). Article CAS PubMed Google
Scholar * Yamaguchi, K. et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. _Science_ 270, 2008–2011 (1995). Article ADS CAS
PubMed Google Scholar * Toma, I. & McCaffrey, T. A. Transforming growth factor-beta and atherosclerosis: interwoven atherogenic and atheroprotective aspects. _Cell Tissue Res_ 347,
155–175 (2012). Article CAS PubMed Google Scholar * Moonen, J. A. J. et al. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by
fluid shear stress. _Cardiovasc Res_ 108, 377–386 (2015). Article ADS CAS PubMed Google Scholar * Wink, D. A. et al. Mechanisms of the antioxidant effects of nitric oxide. _Antioxid
Redox Signal_ 3, 203–213 (2001). Article CAS PubMed Google Scholar * Boon, R. A. et al. KLF2 suppresses TGF-beta signaling in endothelium through induction of Smad7 and inhibition of
AP-1. _Arterioscler Thromb Vasc Biol_ 27, 532–539 (2007). Article CAS PubMed Google Scholar * Guo, Y. et al. Kallistatin inhibits TGF-beta-induced endothelial-mesenchymal transition by
differential regulation of microRNA-21 and eNOS expression. _Exp Cell Res_ 337, 103–110 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Yan, F. et al. Glucagon-like
peptide 1 protects against hyperglycemic-induced endothelial-to-mesenchymal transition and improves myocardial dysfunction by suppressing poly(ADP-ribose) polymerase 1 activity. _Mol Med_
21, 15–25 (2015). Article CAS PubMed PubMed Central Google Scholar * Rassaf, T., Feelisch, M. & Kelm, M. Circulating NO pool: assessment of nitrite and nitroso species in blood and
tissues. _Free Radic Biol Med_ 36, 413–422 (2004). Article CAS PubMed Google Scholar * Clark, P. R. et al. MEK5 is activated by shear stress, activates ERK5 and induces KLF4 to modulate
TNF responses in human dermal microvascular endothelial cells. _Microcirculation_ 18, 102–117 (2011). Article CAS PubMed PubMed Central Google Scholar * Young, A. et al. Flow activation
of AMP-activated protein kinase in vascular endothelium leads to Kruppel-like factor 2 expression. _Arterioscler Thromb Vasc Biol_ 29, 1902–1908 (2009). Article CAS PubMed PubMed Central
Google Scholar * Sakurai, H., Miyoshi, H., Toriumi, W. & Sugita, T. Functional interactions of transforming growth factor beta-activated kinase 1 with IkappaB kinases to stimulate
NF-kappaB activation. _J Biol Chem_ 274, 10641–10648 (1999). Article CAS PubMed Google Scholar * Derynck, R. & Zhang, Y. E. Smad-dependent and Smad-independent pathways in TGF-beta
family signalling. _Nature_ 425, 577–584 (2003). Article ADS CAS PubMed Google Scholar * Sorrentino, A. et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor
kinase-independent manner. _Nat Cell Biol_ 10, 1199–1207 (2008). Article CAS PubMed Google Scholar * Sakurai, H. Targeting of TAK1 in inflammatory disorders and cancer. _Trends Pharmacol
Sci_ 33, 522–530 (2012). Article CAS PubMed Google Scholar * Yan, X., Liu, Z. & Chen, Y. Regulation of TGF-β signaling by Smad7. _Acta Biochim Biophys Sin_ 41, 263–272 (2009).
Article CAS PubMed Google Scholar * Hilgarth, R. S. et al. Regulation and function of SUMO modification. _J Biol Chem_ 279, 53899–53902 (2004). Article CAS PubMed Google Scholar *
Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. _Cell_ 116, 281–297 (2004). Article CAS PubMed Google Scholar * Rassaf, T., Bryan, N. S., Kelm, M. & Feelisch,
M. Concomitant presence of N-nitroso and S-nitroso proteins in human plasma. _Free Radic Biol Med_ 33, 1590–1596 (2002). Article CAS PubMed Google Scholar * Feelisch, M. et al.
Concomitant S-, N-, and heme-nitros(yl)ation in biological tissues and fluids: implications for the fate of NO _in vivo_ . _FASEB J_ 16, 1775–1785 (2002). Article CAS PubMed Google
Scholar * Luscinskas, F. W. et al. Cytokine-activated human endothelial monolayers support enhanced neutrophil transmigration via a mechanism involving both endothelial-leukocyte adhesion
molecule-1 and intercellular adhesion molecule-1. _J Immunol_ 146, 1617–1625 (1991). CAS PubMed Google Scholar * Donker, R. B. et al. Absence of _in vivo_ generalized pro-inflammatory
endothelial activation in severe, early-onset preeclampsia. _J Soc Gynecol Investig_ 12, 518–528 (2005). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank
Mr. Henk E. Moorlag for isolating HUVEC and preparing reagents for endothelial cell culture. We thank Mrs. Rianne M. Jongman and Mrs. Linda A. Brouwer for their technical support. This work
was supported by grants from the Willem Johan Kolff Institute for Biomedical Engineering and Materials Science and Jan-Kornelis de Cock Foundation (De-Cock 11–42, 12-56 & 15-80),
University of Groningen, The Netherlands to E.S. Lee and M.C. Harmsen, as well as by funds from the Faculty of Medicine, University of Southampton, UK to M. Feelisch. TissueFaxs® Microscopy
was performed at the University Medical Center Groningen Microscopy and Imaging Center (UMIC) which is supported by the Netherlands Organisation for Scientific Research (NWO) by grant
175-010-2009-023. The funders had no role in study design, data collection and interpretation, preparation of manuscript or in making decision for publishing the research. AUTHOR INFORMATION
Author notes * Ee Soo Lee Present address: Present address: National University of Singapore, Centre for Life Sciences, Department of Physiology, Singapore, 117456, Singapore., AUTHORS AND
AFFILIATIONS * Department of Pathology and Medical Biology, University of Groningen, University Medical Center Groningen, Groningen, NL-9713, GZ, The Netherlands Ee Soo Lee, Llorenç Solé
Boldo & Martin C. Harmsen * University of Southampton, Southampton General Hospital, Faculty of Medicine, Clinical and Experimental Sciences, Southampton, SO166YD, United Kingdom
Bernadette O. Fernandez & Martin Feelisch Authors * Ee Soo Lee View author publications You can also search for this author inPubMed Google Scholar * Llorenç Solé Boldo View author
publications You can also search for this author inPubMed Google Scholar * Bernadette O. Fernandez View author publications You can also search for this author inPubMed Google Scholar *
Martin Feelisch View author publications You can also search for this author inPubMed Google Scholar * Martin C. Harmsen View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS E.S.L., M.C.H. and M.F. designed the research. E.S.L., L.S.B. and B.O.F. performed experiments. E.S.L. performed data analysis and wrote the article. All
authors reviewed, edited the manuscript and approved its final version. CORRESPONDING AUTHOR Correspondence to Martin C. Harmsen. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare
no competing financial interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION (PDF 927 KB) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0
International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the
material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Lee, E., Boldo, L., Fernandez, B. _et al._ Suppression of TAK1 pathway by shear
stress counteracts the inflammatory endothelial cell phenotype induced by oxidative stress and TGF-β1. _Sci Rep_ 7, 42487 (2017). https://doi.org/10.1038/srep42487 Download citation *
Received: 17 August 2016 * Accepted: 09 January 2017 * Published: 17 February 2017 * DOI: https://doi.org/10.1038/srep42487 SHARE THIS ARTICLE Anyone you share the following link with will
be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative