Evaluation of taqman qpcr system integrating two identically labelled hydrolysis probes in single assay
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Ongoing evolution of viral pathogens is a significant issue in diagnostic virology employing TaqMan qPCR/RT-qPCR. Specific concerns are related to false negativity due to probe
binding failure. One option for compensating for such deficiency is to integrate a second identically labelled probe in the assay. However, how this alteration influences the reaction
parameters has not been comprehensively demonstrated. In the present study, we evaluate a TaqMan protocol using two identically labelled hydrolysis probes (simple, LNA (locked-nucleic-acid))
and MGB (minor-groove-binder) modified probes and combinations thereof in a single assay. Our results based on a synthetic amplicon suggest that the second probe does not compromise the
TaqMan qPCR/RT-qPCR parameters, which repeatedly and reproducibly remained comparable to those of the corresponding single-probe assays, irrespective of the relative probe orientation,
whether opposite or tandem, and probe modifications or combinations thereof. On the other hand, the second probe additively contributed to the overall fluorescence signal. The utility of the
dual-probe approach was demonstrated on practical examples by using field specimens. We hope that the present study might serve as a theoretical basis for the development or improvement of
TaqMan qPCR/RT-qPCR assays for the detection of highly variable nucleic acid templates. SIMILAR CONTENT BEING VIEWED BY OTHERS DEVELOPMENT OF A SENSITIVE, QUANTITATIVE ASSAY WITH BROAD
SUBTYPE SPECIFICITY FOR DETECTION OF TOTAL HIV-1 NUCLEIC ACIDS IN PLASMA AND PBMC Article Open access 28 January 2022 DEVELOPMENT OF LOOP-MEDIATED ISOTHERMAL AMPLIFICATION (LAMP) ASSAYS
USING FIVE PRIMERS REDUCES THE FALSE-POSITIVE RATE IN COVID-19 DIAGNOSIS Article Open access 28 March 2023 DEVELOPMENT AND VALIDATION OF COST-EFFECTIVE ONE-STEP MULTIPLEX RT-PCR ASSAY FOR
DETECTING THE SARS-COV-2 INFECTION USING SYBR GREEN MELTING CURVE ANALYSIS Article Open access 20 April 2022 INTRODUCTION Real-time PCR utilising 5′ nuclease or hydrolysis probes1, also
known as TaqMan qPCR, is an essential and powerful tool used in various areas of life science. It also has great potential in diagnostic microbiology, where the TaqMan qPCR is often the
first-line screening method for the detection of many viral or bacterial pathogens in human, animal or plant specimens. The TaqMan qPCR employs a pair of primers and a non-extendable probe.
The probe is a short, sequence specific oligonucleotide, which binds within the region delimited by the primers. One end, usually the 5′ terminus, of the probe is labelled using a
fluorescent dye, while the other one, the 3′ end, is tagged with a quencher. The fluorescent dye and the quencher form a donor-acceptor FRET (fluorescence resonance energy transfer) pair2.
As the amplification proceeds, the TaqMan probe hybridizes to the target sequence downstream of one of the primers. These two oligonucleotides form an effective unit. Once the upstream
primer begins to be extended, the _Taq_ polymerase mediated 5′→3′ hydrolysis of the probe takes place, leading to the disruption of the FRET pair accompanied by dye release from the
quenching effect. Subsequent illumination of the reaction mix results in detectable fluorescence with a dye-specific wave-length. The emitted fluorescence signal is proportional to the
amount of accumulated PCR product. Conventionally, the TaqMan qPCR is a tri-oligonucleotide system. The probe, as the third oligonucleotide, introduces an additional level of sequence
specificity into the assay. On the other hand, it also substantially contributes to the susceptibility of the technique to template mutations. Mismatches in the probe-binding region decrease
or completely eliminate the probe binding, leading to false negativity. This is an important and frequently reported issue in diagnostic virology3,4,5,6,7,8,9,10,11,12,13,14 that is faced
in the detection of highly variable viral nucleic acid (NA) templates. Therefore, it is crucial to design novel approaches to improve the inclusivity and to reduce the probability of false
negative results of diagnostic TaqMan qPCR assays. One option for improvement comes from the TaqMan principle itself. The parsing of the reaction mechanism in more detail suggests that, in
addition to effective pair-mediated signal generation, the second primer-induced opposite strand synthesis runs in parallel. Since this process is not captured by fluorescence imaging, the
opposite strand remains invisible. Hence, the signal observed during the TaqMan qPCR is generated in fact upon only 50% of the accumulated amplicon strands. Visualization of the “invisible”
amplicon fraction by coupling the second primer with a hydrolysis probe, or more generally by including more distinct probes amidst the forward and reverse primers, could therefore
significantly reduce the probability of probe binding failure and improve the inclusivity of a given assay. Nevertheless, TaqMan RT-qPCR assays utilizing two or more identically or
differently labelled probes15,16,17 are rare in diagnostic microbiology. The probable reason why this approach was not established is the absence of a more comprehensive evaluation
background. Specifically, it is not clear how the identically labelled probes operating alongside one another in a single assay influence the basic reaction parameters, like the Cq,
sensitivity, efficiency and overall fluorescence. Does the second probe alter them? Can we combine identically labelled probes with different modifications like LNA and MGB in the same
assay? Is there a difference between the probes in opposite and tandem orientations? To answer these questions, and to propagate the application of the dual-probe TaqMan approach, in the
present study, we evaluate a TaqMan qPCR protocol using two identically labelled hydrolysis probes binding to discrete, non-overlapping template regions in a single qPCR assay. Based on a
synthetic DNA template construct, we investigate the effect of the dual-probe assay on the basic reaction parameters, including the Cq, relative fluorescence intensity, efficiency,
repeatability, reproducibility and sensitivity. All of the parameters were evaluated for simple (unmodified) probes, LNA or MGB modified probes, and combinations thereof either in opposite
or tandem orientations. Finally, the utility of the presented approach in diagnostic qPCR was demonstrated on practical examples. RESULTS NUCLEIC ACID STANDARD DESIGN Theoretically, there
are two options for how to integrate a second hydrolysis probe into the TaqMan assay: (i) in opposite orientation, i.e. downstream of the second primer, thus creating a second effective unit
(primer-probe pair), the two units then each bind on one template strand; or (ii) in tandem or serial orientation, where the probes bind consecutively downstream of one of the primers. To
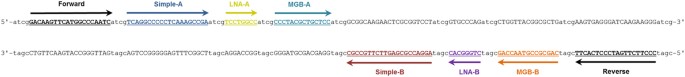
test both options, a synthetic NA standard was designed (Fig. 1). The standard sequence starts with a short tag, followed by a pair of primers delimiting the amplicon. The amplicon
encompasses six probe-binding motifs, three located downstream of the forward primer and three located downstream of the reverse primer. The probe sequences (Fig. 2) were selected from the
previously published assays so as to enable the testing of simple and LNA and MGB modified probes in various combinations, all labelled with a FAM reporter dye. So, each consecutive triplet
contains one simple (Simple-A18, Simple-B19), one LNA modified (LNA-A20, LNA-B21), and one MGB modified (MGB-A22, MGB-B23) probe motif. The designed NA standard sequence enabled nine
different assays with the probes in opposite orientations to be set up: 1. simple-simple, 2. LNA-LNA, 3. MGB-MGB, and simple-LNA, simple-MGB, and LNA-MGB pairs, each in two permutations. In
addition, it was possible to set up six probes in tandem orientations (simple-LNA, simple-MGB, and LNA-MGB, each in two permutations). This provided numerous testing opportunities, with only
the combinations most likely to be applied being further evaluated. EVALUATION OF REACTION PARAMETERS OF DUAL-PROBE TAQMAN QPCR ASSAY Six combinations of probes in opposite orientation: 1.
simple-simple, 2. simple-MGB, 3. simple-LNA (Fig. 3 Panels a–c) and 4. MGB-MGB, 5. LNA-LNA, and 6. LNA-MGB (Supplementary Information 1, Fig. S1.1, Panels a–c), were selected to test the
opposite dual-probe qPCR assay. The tandem orientations were investigated in three probe combinations: 1. simple-MGB, 2. simple-LNA, and 3. LNA-MGB (Supplementary Information 2, Fig. S2.1,
Panels a–c). Dilution and calibration curves covering a quantitative range of six magnitudes (from 1e2 to 1e7 NA standard copies per μl of template), were prepared to evaluate the reaction
parameters. The dilution curves showed a parallel course for the particular dual-probe and the corresponding single-probe subsets in the early exponential phase of amplification. This
resulted in highly similar Cq values, supported by low standard deviation (SD) across the entire dilution gradient, irrespective of the combination of the hydrolysis probe modifications or
the tested orientations thereof. The close Cq values were also reflected in the parallel and overlapping calibration curves, where the corresponding assays had comparable amplification
efficiencies (Fig. 4, Panels a–c and Supplementary Information 1, Fig. S1.2, Panels a–c, and Supplementary Information 2, Fig. S2.2, Panels a–c). Similarly, the ΔCq straight lines showed
parallel courses, with the slope values significantly below the 0.1 threshold (Fig. 4, Panels d–f and Supplementary Information 1, Fig. S1.2, Panels d–f and Supplementary Information 2, Fig.
S2.2, Panels d–f), again suggesting that the dual-probe and the corresponding single-probe TaqMan assays have comparable efficiencies. One slight deviation in the data was observed. In
particular, the opposite MGB-A+LNA-B assay (Supplementary Information 1, Fig. S1.2, Panel c) showed slightly lower efficiency than the corresponding MGB-A subset. This was also reflected in
the elevated ΔCq slope of 0.157 (Supplementary Information 1, Fig. S1.2, Panel f), which, though small, is still higher than the 0.1 threshold. However, the observed deviation was attributed
to experimental imperfections and did not contradict the general findings of the study. Taken together, the calibration curve and ΔCq slope data clearly proved that the second identically
labelled probe did not significantly influence the TaqMan qPCR assay. The dual-probe assays stably showed highly similar parameters, in comparison with the single-probe counterparts,
independently of the probe modifications, orientations or combinations tested. EVALUATION OF REACTION FLUORESCENCE OF DUAL-PROBE TAQMAN QPCR ASSAY As was expected, the second probe in the
TaqMan qPCR assay had an additive effect on reaction fluorescence. To quantitatively express the observed differences, the steepness and the first derivative maxima of the amplification
curves were compared between the corresponding subsets across the entire dilution gradient (Fig. 3, Panels d–f and Supplementary Information 1, Fig. S1.1, Panels d–f and Supplementary
Information 2, Fig. S2.1, Panels d–f). The comparison revealed that the opposite probe assays and the tandem dual-probe assays had both steeper and brighter amplification curves (15 to 60%
and 20 to 43%, respectively) than the corresponding single-probe counterparts (Supplementary Information 1, Table S1.1 and Supplementary Information 1, Table S2.1). The observed differences
in fluorescence were retained across the entire dilution gradient, regardless of the probe combination tested. REPEATABILITY AND REPRODUCIBILITY OF DUAL-PROBE TAQMAN QPCR ASSAY The
intra-assay variation (i.e. repeatability) was tested on three different concentrations (high, 1e6; medium, 1e4; and low, 1e2 NA standard copies per μl of template, each in triplicate) in a
single run. Evaluation of the mean Cq and SD of the particular standard triplicates showed SD values to be lower than or equal to 0.41 for all six opposite dual-probe TaqMan assays
(Supplementary Information 1, Table S1.2). The three tandem-probe assays exhibited SD ≤ 0.56. These results indicated low spread from the mean and thus low intra-assay variation within all
investigated runs (Supplementary Information 2, Table S2.2). The inter-assay variation (i.e. reproducibility) was tested by three operators on three CFX 96 instruments using that same
standard set. The mean Cq, SD, and %RSD values were as follows: SD ≤ 0.67 and %RSD ≤ 2.36% for the opposite dual-probe TaqMan assays and SD ≤ 0.52 and %RSD ≤ 1.60% for the tandem-probe
assays (Supplementary Information 1, Table S1.3 and Supplementary Information 2, Table S2.3). The results proved the high degree of precision of the data and low inter-assay variation.
SENSITIVITY OF DUAL-PROBE TAQMAN QPCR ASSAY The results of the sensitivity analyses are summarised in Fig. 5a and b. Overall, the ΔCq scatter plot, including 2 × 42 paired analyses for all
opposite probe combinations, showed predominant distribution of the ΔCq values below the diagonal, or more specifically, within the interval of 0 and −2 (dual-probe mean ΔCq 35.58 ± 1.89
versus 36.19 ± 1.91 and 35.97 ± 2.01). The tandem dual-probe assays, represented by 2 × 18 paired analyses exhibited symmetrical distribution of the particular ΔCq values across the diagonal
(dual-probe mean ΔCq 35.28 ± 1.80 versus 35.52 ± 2.05 and 35.28 ± 1.93). The results sufficiently proved that including the second identically labelled probe into the TaqMan qPCR does not
compromise the assay sensitivity, with Cq values remaining virtually identical, even at limited template concentrations. This was observed irrespective of the probe modification, combination
or orientation. Finally, the influence of the second probe on the FAM signal and data interpretation at limited standard concentrations was investigated. The %d(RFU) scatter plots (see
methods) consistently expressed stronger FAM signal across the entire panel, with the data distribution between 10–60% for all tested probe combinations. DEMONSTRATION OF UTILITY OF
DUAL-PROBE TAQMAN QPCR IN DIAGNOSTIC MICROBIOLOGY The utility of the dual-probe approach in improving the diagnostic TaqMan qPCR was demonstrated on two assays: Equine arteritis virus (EAV)
RT-qPCR and Canine parvovirus 2 (CPV-2) qPCR (Supplementary Information 3, Table S3.1). The EAV assay used simple-simple probes in opposite orientation, while the CPV-2 assay utilized a
simple-MGB tandem-probe pair. To investigate the mutual compensatory effect of the probe pairs during the amplification, various EAV and CPV-2 strains holding different nucleotide mutations
that weakened or knocked out one of the probes were selected. The most striking examples were shown in Fig. 6. A dual-probe (tandem simple-MGB configuration) CPV-2 qPCR and the corresponding
single-probe assays were used in triplicates to investigate a CPV-2 strain, which perfectly matched the first probe but held three nucleotide changes in the second probe binding site. One
of these changes was located approximately in the middle, while the last two were situated close to the MGB-modified 3′ end of the probe. The results revealed that the observed mutations
were deleterious to the CPV-2_probe2 binding and hydrolysis. Hence, the CPV-2_probe2 qPCR assay failed to detect this CPV-2 strain despite the presence of the underlying amplicon, as was
suggested by electrophoresis. On the contrary, the dual-probe assay showed clear CPV-2 positivity with virtually identical parameters to the CPV-2_probe1 counterpart. This indicated that in
the dual-probe setting, the CPV-2_probe1 sufficiently compensated for the CPV-2_probe2 deficiency. In the second example, a dual-probe EAV RT-qPCR assay (opposite simple-simple
configuration) was used, along with the single-probe counterparts, to detect an EAV strain with 100% similarity to the EAV_probe2, while carrying two mutations relative to the EAV_probe1.
The first of these mutations was located at the extreme 5′ end, suggesting possible interference with hydrolysis. The second change was slightly eccentric and shifted towards the 3′ end.
Interestingly, as observed on the amplification plot, the EAV_probe2 probe was still capable of detecting the tested EAV strain, but with weak FAM signal gain resulting in a flat
amplification curve. Nevertheless, even this weak fluorescence yield can contribute to the overall signal of the dual-probe assay, which outperformed the perfectly matching single-probe
assay. Additional examples of the dual-probe CPV-2 and EAV assays were provided in Supplementary Information 3, Figure S3a–c. DISCUSSION Ongoing evolution of viral pathogens is a significant
issue in diagnostic virology employing TaqMan qPCR/RT-qPCR. Specific concerns are related to false negativity as a result of decreased probe binding efficiency or complete binding failure
due to mutations in the corresponding template regions3,4,5,6,7,8,9,10,11,12,13,14. Hence, even if the specific amplicons are generated, the assays remain non-fluorescent, thus suggesting no
amplification in a TaqMan qPCR setting. Including a second probe in the assay may therefore compensate for the deficiency of the first one and restore the assay inclusivity. There are
several options for how to integrate the second probe into the TaqMan qPCR assay. The probes can bind to the complementary amplicon strand or consecutively downstream of one of the primers.
Further, they can be simple or additionally modified to increase the melting temperature. Finally, the probes can hold identical or different fluorescent labels. Our results, based on a
synthetic NA construct, suggested that the integration of two identically labelled hydrolysis probes in an equimolar ratio does not compromise the basic parameters of the TaqMan
qPCR/RT-qPCR. The dual-probe assays repeatedly and reproducibly retained comparable Cq, reaction efficiency and sensitivity to the corresponding single-probe counterparts. In addition, these
parameters were not influenced by the relative probe orientations, whether opposite or tandem, nor were they affected by probe modifications or combinations. The capacity of the dual-probes
to mutually complement one another in real conditions was demonstrated on two distinct assays, CPV-2 qPCR with tandem simple-MGB, and EAV RT-qPCR with opposite simple-simple configurations.
The templates were CPV-2 and EAV positive field specimens with various mismatches in one of the probe binding regions, which weakened or completely eliminated the hybridization.
Unfortunately, we had no viral strains in our repository holding partially deleterious mutations in both of the probe binding sites. Hence, the effect of such mutations remains to be
elucidated. As was shown, the perfectly matched probes were able to fully compensate for the decreased efficiency or the complete failure of the second one. In spite of this, the weakened
probes were able to contribute to the overall fluorescence signal, which facilitates the assay evaluation. All of these conclusions agreed with our initial conclusions based on the synthetic
DNA construct. However, we would like to emphasize that the CPV-2 and EAV assays were set up for demonstration purposes only, and their use for routine screening would require optimization
and comprehensive validation, including a broad panel of negative specimens to exclude interference between the probes and generation of false positive signals. Although the general results
of our study agreed well with the previous findings of Yip and colleagues15, the Cq and efficiency values of the dual-probe assays are slightly contradictory. Specifically, we did not find
as significant improvement in Cq and efficiency values of the dual-probe assays across our data set as reported by Yip and colleagues15. However, the theory of qPCR does not suggest
significant improvements in these parameters either. In tandem orientations, the two probes bind to one of the DNA template strands. Assuming that the tandem-probe and the corresponding
single-probe assays exhibit comparable efficiencies, the recognised template amount in each cycle is identical, i.e. 50%, since in both cases the probe binding and hydrolysis take place upon
a single strand of a double-strand template. Hence, all the assays would have identical Cq values and the differences would only be reflected in the higher fluorescence yield of the
tandem-probe assays. On the contrary, a dual-probe assay with opposite probe configuration simultaneously detects both strands of the template during each single cycle. Indeed, there is a
2-fold difference between the opposite and tandem-probe assays, which would theoretically yield lower Cq values. Nevertheless, the theory further suggests that if a qPCR is 100% efficient,
the 2-fold measurements differ by one cycle between the means24. However, in reality, the amplification efficiency is never absolute, and therefore, the expected one cycle difference between
the 2-fold dilutions is reduced. In addition, it is burdened with higher experimental variations25. Hence, the Cq improving-effect of the second probe in opposite orientation is difficult
to distinguish. In summary, there is no obvious theoretical explanation for the observed contradictions. Instead, it seems that the Cq and reaction efficiency improvements observed by Yip
and colleagues15 resulted from sequence mismatches in the first probe and the compensatory effect of the second one. This option was not further investigated. On the other hand, the
dual-probe approach greatly improved the reaction fluorescence. In agreement with Yip and colleagues15, the second identically labelled probe had an additive effect on the overall reaction
fluorescence, which increased it by up to 60% in comparison with the single-probe assays. This represents a significant improvement which facilitates the evaluation and interpretation of
weak positive specimens. Further, identically labelled FAM probes enable pre-existing internal controls to be used without the necessity of re-labelling. Finally, they provide the option of
advanced analyses14, and their use is cost-effective. However, the major advantage of the dual-probe approach does not lie in the improvement of the reaction parameters itself, but in the
ability to increase the inclusivity of the diagnostic TaqMan assay. The second probe would extend or restore the potential of the pre-existing diagnostic assays to detect novel genetic
variants. This potential is of utmost importance in universal assays, which are often used as the first-line screening tool in diagnostic microbiology20,26,27,28. Universality should also be
kept in mind during the development of any novel TaqMan screening assay. To meet this requirement, the primers and the probes have to be selected at the most conserved regions of the
targeted genome. Optionally, evolutionarily conserved regions are identified by multiple sequence alignment (MSA), including sequences from a broad time period and from different geographic
regions and host species. However, the more diverse the sequence collection of the MSA, the less abundant the conserved positions are. So, once the primers have been selected, the MSA may
not provide additional evolutionarily conserved positions suitable for probe selection. The advancement of the dual-probe TaqMan strategy is to enable probe selection at semi-conserved
motifs, where the two probes may complement or overlap one another in sequence inclusivity. This might facilitate the development of novel screening assays. It is worth bearing in mind,
however, that the opposite dual-probe system is inherently predisposed towards lower Cq values and should be the preferred choice during assay design. Despite the indisputable advantages,
the dual-probe TaqMan technique has several drawbacks. Primarily, the second probe introduces an additional level of complexity into the assay. Therefore, the dual-probe assay requires more
comprehensive optimization, evaluation and validation than a single-probe TaqMan assay. Attention should mainly be paid to the generation of nonspecific fluorescent signals, which should be
tested on a panel of negative specimens, as well as non-template controls. Further, when one of the probes is knocked out, it cannot be distinguished at first glance which one was
influenced. The probe failure is reflected only in the drop of the overall signal intensity of the assay. This requires more attention during the interpretation of the results. Additional
disadvantages are the increased reaction cost and possibly an extra pipetting step during the master mix preparation. However, in spite of the negative aspects mentioned above, we are firmly
convinced that the dual-probe strategy using identically labelled probes may serve as a valuable option in the development or improvement of a TaqMan qPCR/RT-qPCR assay for the detection of
highly variable nucleic acid templates. METHODS FIELD SPECIMENS Canine parvovirus 2 (CPV-2) positive canine organ suspensions and Equine arteritis virus (EAV) positive equine semen
specimens were selected for analysis. None of the specimens were initially collected for the purpose of this study. The ethical standards in animal welfare and protection are subject to
inspection by the State Veterinary Administration of the Czech Republic. NUCLEIC ACID EXTRACTION Total NA was extracted using the MagNAPure Compact (Total NA Extraction Kit I, Roche), with
input sample volumes from 200 or 400 μl and elution volume of 50 μl. NUCLEIC ACID STANDARD A synthetic DNA construct (Fig. 1, Integrated DNA Technologies) in a 4 nM scale was diluted in 1 ml
of nuclease free water (Qiagen). The copy number/μl of the stock was then calculated and used for downstream experiments. The primers and unmodified dually labelled FAM-BHQ1 hydrolysis
probes (Fig. 2) were supplied by Generi Biotech, Czech Republic. The MGB probes (Life Technologies) incorporated a FAM reporter dye and a 3′ nonfluorescent quencher (NFQ), with the MGB
moiety attached to the quencher molecule. The LNA probes, UPL#104 and UPL#162, were FAM and dark quencher dye (DDQ) labelled octamers selected from the 165 Universal Probe Library (Roche)29.
DUAL-PROBE TAQMAN QPCR All of the reaction mixes were prepared using QuantiTect Probe PCR or QuantiTect Probe RT-PCR Kits (Qiagen), with a 0.6 μM of primers and 0.2 μM of each probe in a
final volume of 12.5 μl (10.5 μl reaction mix and 2 μl of DNA template), using white opaque and foil-sealed plates (Bioplastics). The primers and the probe sequences for the DNA standard are
listed in Fig. 2. The qPCR thermoprofiles for the synthetic DNA construct universally started with an initial activation at 95 °C for 15 min, followed by 45 cycles of 95 °C for 10 s and 60
°C for 30 s with a signal acquisition in the FAM channel at the end of the annealing/extension step. The dual-probe CPV-2 qPCR assay consisted of a tandem simple-MGB probes, amplifying a 340
bp-long amplicon with the forward primer and first probe according to Streck end colleagues30, and the reverse primer and the second probe according to Decaro end colleagues31. The
thermo-profile started at 95 °C for 15 min, followed by 45 cycles of 95 °C for 10 s and 60 °C for 1 min, with a signal acquisition in the FAM channel. The dual-probe EAV RT-qPCR assay
delimited a 399 bp-long region32 and included a pair of simple probes32,33 in opposite orientation. The thermo-profile started with reverse transcription of 50 °C for 30 min and 95 °C for 15
min, followed by 45 cycles of 95 °C for 20 s, 60 °C for 1 min and 72 °C for 30 s, with a signal acquisition in the FAM channel at the end of the annealing/extension step. The primers and
the probe sequences for the CPV-2 and EAV assays are listed in Supplementary Information 3, Table 3.1. All of the reactions were run in triplicates on a CFX 96 (BioRad) thermal cycler. The
Cq values were estimated by analysing the data as a single pool, using the automatic threshold and baseline cycle option of the CFX Manager Software v3.1. The reactions of interest were
subjected to electrophoretic analysis by using ethidium bromide-containing 2% agarose gel in a TAE buffer and 10 V/cm. SEQUENCE ANALYSIS The amplicons of interest were purified by using a
High Pure PCR Purification Kit (Roche). Sequencing reactions were prepared using a Big Dye Terminator Cycle Sequencing Kit v3.1 and evaluated on 3130 or 3500 Genetic Analysers (all from Life
Technologies). The sequences were aligned manually. The graphical alignment was generated in a BioEdit sequence alignment editor34. EVALUATION OF REACTION PARAMETERS OF DUAL-PROBE TAQMAN
QPCR ASSAY Dilution and calibration curves were constructed on the basis of a 10-fold dilution gradient of the synthetic DNA standard, covering a quantitative range of six magnitudes (from
1e2 to 1e7 NA standard copies per μl of template) in triplicates including three non-template controls. To evaluate the effect of the second probe on the TaqMan reaction, each individual run
was set up in three subsets. The first subset represented the dual-probe reaction (0.2 μM of each probe), while the second and third ones contained the corresponding single probes (0.2 μM)
in separate reactions. For each subset, the efficiency E, slope _k,_ and the equation and R2 values of the calibration curve regression line were determined. To infer the comparability of
the two probe assays with the single-probe counterparts, the ΔCq slope approach was used35. First, the ΔCq between the dual-probe and corresponding single-probe reactions was calculated
according to the formula: ΔCq = Cq(dual-probe) - Cq(simple-probe), and plotted against the logarithm of the template concentration. Then, the slopes of the resulting straight lines were
compared between the reactions. If the slope values were <0.1, the reaction efficiencies were comparable. EVALUATION OF REACTION FLUORESCENCE OF DUAL-PROBE TAQMAN QPCR ASSAY The relative
fluorescence unit (RFU) values of the amplification curves gathered in the FAM channel were compared between the subsets. We defined two parameters: the d(RFU) and the steepness _k_. The
d(RFU) represents the fluorescence value at a specific point of the sigmoidal trajectory, which corresponds with its first derivative maximum. The steepness was defined as the slope of the
linear regression line driven along the exponential region of the amplification curve. The exponential region was determined from those RFU data points that provided the highest correlation
(R2 > 0.995) of the regression line. REPEATABILITY AND REPRODUCIBILITY OF DUAL-PROBE TAQMAN QPCR ASSAY The repeatability or intra-assay variation of the two probe TaqMan assays was
estimated from the mean Cq and standard deviation (SD) values obtained from three dilutions of the NA standard, 1e2, 1e4, and 1e6 copies per μl of template in three replicates in a single
run. The reproducibility, i.e. inter-assay variation of the two probe TaqMan assays was inferred from three independent runs of the above three NA standard dilutions prepared by three
distinct operators and run on three different CFX 96 instruments. Then, the mean Cq, SD, and relative standard deviation as a percentage (%RSD, also known as the coefficient of variation)
were calculated. SENSITIVITY OF DUAL-PROBE TAQMAN QPCR ASSAY The relative sensitivity of the dual-probe TaqMan qPCR assays was tested on six opposite probe and three tandem-probe assays by
using various NA standard dilutions to reach Cq values ≥ 30. The particular ΔCq values between the corresponding dual-probe and single-probe assays were estimated according to the above
formula and compared. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Nagy, A. _et al_. Evaluation of TaqMan qPCR System Integrating Two Identically Labelled Hydrolysis Probes in Single
Assay. _Sci. Rep._ 7, 41392; doi: 10.1038/srep41392 (2017). PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. REFERENCES * Holland, P. M., Abramson, R. D., Watson, R. & Gelfand, D. H. Detection of specific polymerase chain reaction product by utilizing the 5′–3′ exonuclease
activity of Thermus aquaticus DNA polymerase. _Proc. Natl. Acad. Sci. USA_ 88, 7276–7280 (1991). Article CAS ADS PubMed Google Scholar * Behlke, M. A., Huang, L., Bogh, L., Rose, S.
& Devor, E. Fluorescence and fluorescence applications. _Integrated DNA Technologies_. (2005). * Stevenson, J., Hymas, W. & Hillyard, D. Effect of sequence polymorphisms on
performance of two real-time PCR assays for detection of herpes simplex virus. _J. Clin. Microbiol._ 43, 2391–2398 (2005). Article CAS PubMed PubMed Central Google Scholar * Kim, L. M.,
Afonso, C. L. & Suarez, D. L. Effect of probe-site mismatches on detection of virulent Newcastle disease viruses using a fusion-gene real-time reverse transcription polymerase chain
reaction test. _J. Vet. Diagn. Invest._ 18, 519–28 (2006). Article PubMed Google Scholar * Lengerova, M. et al. Real-time PCR diagnostics failure caused by nucleotide variability within
exon 4 of the human cytomegalovirus major immediate-early gene. _J. Clin. Microbiol._ 45, 1042–4 (2007). Article CAS PubMed PubMed Central Google Scholar * Lemmon, G. H. & Gardner,
S. N. Predicting the sensitivity and specificity of published real-time PCR assays. _Ann. Clin. Microbiol. Antimicrob_. 7, 18 (2008). Article PubMed PubMed Central Google Scholar *
Cattoli, G. et al. False-negative results of a validated real-time PCR protocol for diagnosis of newcastle disease due to genetic variability of the matrix gene. _J. Clin. Microbiol._ 47,
3791–3792 (2009). Article CAS PubMed PubMed Central Google Scholar * Klungthong, C. et al. The impact of primer and probe-template mismatches on the sensitivity of pandemic influenza
A/H1N1/2009 virus detection by real-time RT-PCR. _J Clin Virol_. 48, 91–95 (2010). Article CAS PubMed Google Scholar * Lee, H. K. et al. Missed diagnosis of influenza B virus due to
nucleoprotein sequence mutations, Singapore. _Euro Surveill_. 16, pii19943 (2011). Google Scholar * Armstrong, P. M., Prince, N. & Andreadis, T. G. Development of a multi-target TaqMan
assay to detect eastern equine encephalitis virus variants in mosquitoes. _Vector Borne Zoonotic Dis._ 12, 872–876 (2012). Article PubMed PubMed Central Google Scholar * Brault, A. C.,
Fang, Y., Dannen, M., Anishchenko, M. & Reisen, W. K. A naturally occurring mutation within the probe-binding region compromises a molecular-based West Nile virus surveillance assay for
mosquito pools (Diptera: Culicidae). _J. Med. Entomol._ 49, 939–41 (2012). Article PubMed PubMed Central Google Scholar * Garson, J. A. et al. Minor groove binder modification of widely
used TaqMan probe for hepatitis E virus reduces risk of false negative real-time PCR results. _J. Virol. Methods._ 186, 157–160 (2012). Article CAS PubMed Google Scholar * Steensels, D.,
Vankeerberghen, A. & De Beenhouwer, H. Towards multitarget testing in molecular microbiology. _Int. J. Microbiol_. ID121057 (2013). * Nagy, A. et al. MeltMan: Optimization, Evaluation,
and Universal Application of a qPCR System Integrating the TaqMan qPCR and Melting Analysis into a Single Assay. _PLoS One._ 11, e0151204 (2016). Article PubMed PubMed Central Google
Scholar * Yip, S. P. et al. Use of dual TaqMan probes to increase the sensitivity of 1-step quantitative reverse transcription-PCR: application to the detection of SARS coronavirus. _Clin.
Chem_. 51, 1885–8 (2005). Article CAS PubMed Google Scholar * Aitichou, M. et al. Dual-probe real-time PCR assay for detection of variola or other orthopoxviruses with dried reagents.
_J. Virol. Methods._ 153, 190–5 (2008). Article CAS PubMed Google Scholar * Zhao, Y. et al. Locked Nucleic Acid Probe-Based Real-Time PCR Assay for the Rapid Detection of
Rifampin-Resistant Mycobacterium tuberculosis. _PLoS One._ 10, e0143444 (2015). Article PubMed PubMed Central Google Scholar * Spackman, E. et al. Development of a real-time reverse
transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. _J. Clin. Microbiol._ 40, 3256–3260 (2002). Article CAS PubMed PubMed Central Google
Scholar * Abril, C. et al. Both viral and host factors contribute to neurovirulence of bovine herpesviruses 1 and 5 in interferon receptor-deficient mice. _J. Virol._ 78, 3644–3653 (2004).
Article CAS PubMed PubMed Central Google Scholar * Nagy, A. et al. Development and evaluation of a one-step real-time RT-PCR assay for universal detection of influenza A viruses from
avian and mammal species. _Arch. Virol._ 155, 665–73 (2010). Article CAS PubMed Google Scholar * Fernández-Pinero, J. et al. Molecular diagnosis of African Swine Fever by a new real-time
PCR using universal probe library. _Transbound. Emerg. Dis_. 60, 48–58 (2013). Article PubMed Google Scholar * Diallo, I. S., Hewitson, G., Wright, L., Rodwell, B. J. & Corney, B. G.
Detection of equine herpesvirus type 1 using a real-time polymerase chain reaction. _J. Virol. Methods._ 131, 92–8 (2006). Article CAS PubMed Google Scholar * Diallo, I. S. et al.
Multiplex real-time PCR for the detection and differentiation of equid herpesvirus 1 (EHV-1) and equid herpesvirus 4 (EHV-4). _Vet. Microbiol._ 123, 93–103 (2007). Article CAS PubMed
Google Scholar * Real-time PCR: understanding Ct. ThermoFisher Scientific (2016). * Amplification Efficiency of TaqMan Gene Expression Assays. Applied Biosystems (2004). * Fuller, C. M.,
Brodd, L., Irvine, R. M., Alexander, D. J. & Aldous, E. W. Development of an L gene real-time reverse-transcription PCR assay for the detection of avian paramyxovirus type 1 RNA in
clinical samples. _Arch. Virol._ 155, 817–23 (2010). Article CAS PubMed Google Scholar * Hayman, D. T. et al. A universal real-time assay for the detection of Lyssaviruses. _J. Viro.l
Methods_. 177, 87–93 (2011). Article CAS Google Scholar * Alm, E. Universal single-probe RT-PCR assay for diagnosis of dengue virus infections. _PLoS. Negl. Trop. Dis_. 8, e3416 (2014).
Article PubMed PubMed Central Google Scholar * U. P. L., Universal Probe Library, Roche. Available at: https://lifescience.roche.com/webapp/wcs/stores/servlet/Product2_15006_10001_68068
(Accessed: 5th May 2016). * Streck, A. F., Rüster, D., Truyen, U. & Homeier, T. An updated TaqMan real-time PCR for canine and feline parvoviruses. _J. Virol. Methods._ 193, 6–8 (2013).
Article CAS PubMed Google Scholar * Decaro, N. et al. Specific identification of feline panleukopenia virus and its rapid differentiation from canine parvoviruses using minor groove
binder probes. _J. Virol. Methods._ 147, 67–71 (2008). Article CAS PubMed Google Scholar * Westcott, D. G. et al. Use of an internal standard in a closed one-tube RT-PCR for the
detection of equine arteritis virus RNA with fluorescent probes. _Vet. Res._ 34, 165–76 (2003). Article CAS PubMed Google Scholar * Balasuriya, U. B. et al. Detection of equine arteritis
virus by real-time TaqMan reverse transcription-PCR assay. _J. Virol. Methods._ 101, 21–8 (2002). Article CAS PubMed Google Scholar * Hall, T. A. BioEdit: a user-friendly biological
sequence alignment editor and analysis program for Windows 95/98/NT. _Nucl. Acids. Symp. Ser._ 41, 95–98 (1999). CAS Google Scholar * Critical factors for successful real-time PCR. Qiagen.
_Real-Time PCR Brochure._ 07, 1–63 (2010). Download references ACKNOWLEDGEMENTS This work was supported by the Ministry of Health, Czech Republic-conceptual development of research
organization (National Institute of Public Health-NIPH, IN 75010330). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * State Veterinary Institute Prague, Laboratory of Molecular Methods,
Prague, 16503, Czech Republic Alexander Nagy, Eliška Vitásková, Lenka Černíková & Vlastimil Křivda * National Institute of Public Health, National Reference Laboratory for Influenza,
Prague, Czech Republic Alexander Nagy, Helena Jiřincová & Martina Havlíčková * Department of Virology and Serology, State Veterinary Institute Prague, Prague, 16503, Czech Republic
Vlastimil Křivda, Kamil Sedlák & Jitka Horníčková Authors * Alexander Nagy View author publications You can also search for this author inPubMed Google Scholar * Eliška Vitásková View
author publications You can also search for this author inPubMed Google Scholar * Lenka Černíková View author publications You can also search for this author inPubMed Google Scholar *
Vlastimil Křivda View author publications You can also search for this author inPubMed Google Scholar * Helena Jiřincová View author publications You can also search for this author inPubMed
Google Scholar * Kamil Sedlák View author publications You can also search for this author inPubMed Google Scholar * Jitka Horníčková View author publications You can also search for this
author inPubMed Google Scholar * Martina Havlíčková View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS A.N. conceived the experiments and
wrote the manuscript. A.N., L.Č., E.V. and V.K. conducted the experiments and evaluated the experimental data. H.J., K.S., J.H. and M.H. analysed the results and reviewed the manuscript.
CORRESPONDING AUTHOR Correspondence to Alexander Nagy. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
INFORMATION (PDF 2124 KB) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this
article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will
need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Nagy, A., Vitásková, E., Černíková, L. _et al._ Evaluation of TaqMan qPCR System Integrating Two Identically Labelled Hydrolysis Probes in Single Assay. _Sci
Rep_ 7, 41392 (2017). https://doi.org/10.1038/srep41392 Download citation * Received: 24 August 2016 * Accepted: 14 December 2016 * Published: 25 January 2017 * DOI:
https://doi.org/10.1038/srep41392 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently
available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative