Brain metastasis dna methylomes, a novel resource for the identification of biological and clinical features
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Brain metastases (BM) are one the most lethal and poorly managed clinical complications in cancer patients. These secondary tumors represent the most common intracranial neoplasm in
adults, most frequently originating from lung cancer, breast cancer, and cutaneous melanoma. In primary brain tumors, such as gliomas, recent advances in DNA methylation profiling have
allowed for a comprehensive molecular classification. Such data provide prognostic information, in addition to helping predict patient response to specific systemic therapies. However,
epigenetic alterations of metastatic brain tumors with specific biological and translational relevance still require much further exploration. Using the widely employed Illumina Infinium
HumanMethylation 450K platform, we have generated a cohort of genome-wide DNA methylomes from ninety-six needle-dissected BM specimens from patients with lung cancer, breast cancer, and
cutaneous melanoma with clinical, pathological, and demographic annotations. This resource offers an unprecedented and unique opportunity to identify novel DNA methylation features
influencing the behavior of brain metastasis, and thus accelerate the discovery of BM-specific theranostic epigenetic alterations. Design Type(s) disease state design • DNA methylation
profiling by array design Measurement Type(s) DNA residue methylation Technology Type(s) DNA methylation profiling assay Factor Type(s) Primary Malignant Neoplasm • Tumor Subtype Sample
Characteristic(s) Homo sapiens • brain Machine-accessible metadata file describing the reported data (ISA-Tab format) SIMILAR CONTENT BEING VIEWED BY OTHERS PREDICTION OF BRAIN METASTASIS
DEVELOPMENT WITH DNA METHYLATION SIGNATURES Article Open access 08 October 2024 GENOME-WIDE METHYLATION ANALYSES IDENTIFIES NON-CODING RNA GENES DYSREGULATED IN BREAST TUMOURS THAT
METASTASISE TO THE BRAIN Article Open access 20 January 2022 THE CELL-FREE DNA METHYLOME CAPTURES DISTINCTIONS BETWEEN LOCALIZED AND METASTATIC PROSTATE TUMORS Article Open access 29 October
2022 BACKGROUND & SUMMARY Patients diagnosed with brain metastasis (BM) have a poor quality of life and a dismal prognosis, with survival ranging from three to 25 months1,2. While
developments in systemic drug treatments have significantly improved the survival of patients with extracranial metastases, BM lesions have shown limited response rates to these approaches3.
This trend, along with improvements in neuro-diagnostic imaging techniques, has resulted in an increased incidence of metastatic brain tumors4. In large epidemiological studies, 8–10% of
patients with solid tumors develop BM, this number increases up to 26% when brain autopsy studies are performed5–9. In fact, BM represents the most common intracranial neoplasm in adults,
outnumbering even primary brain tumors. Interestingly, reflecting an organotropic behavior of tumor cells, the vast majority of secondary brain neoplasms (75-90%) are originated in patients
with lung cancer, breast cancer, and cutaneous melanoma5–8. Genomic and epigenomic landscapes of primary brain tumors have been extensively investigated. In gliomas, for example, the
cytosine-guanine island (CGI) methylator phenotype (CIMP) is frequently found in patients with lower grade gliomas harboring mutations on the _IDH1_ gene and is significantly associated with
a better overall prognosis10. Additially, a favorable response to the DNA alkylating antineoplastic agent, temozolomide, has been directly connected to high DNA methylation (DNAm) level in
the promoter region of the _MGMT_ gene11. DNAm profiling has recently been shown to have the potential to accurately stratify primary central nervous system (CNS) tumours12 and to
significantly improve the diagnosis of cancer of unknown primary13. These clinically relevant findings have demonstrated DNAm profiling to be a valuable tool in the histomolecular evaluation
of brain tumors14,15. Yet, while genomic and transcriptomic characterization has been performed to some extent16, clinically relevant epigenetic alterations of metastatic brain tumors are
still poorly understood. Therefore, given this significant knowledge gap, we constructed a comprehensive dataset that can be used to accelerate the identification of novel DNAm features with
biological and clinical relevance for the three most frequent types of BM. Here, we present a dataset including genome-wide DNA methylomes constructed using Illumina Infinium
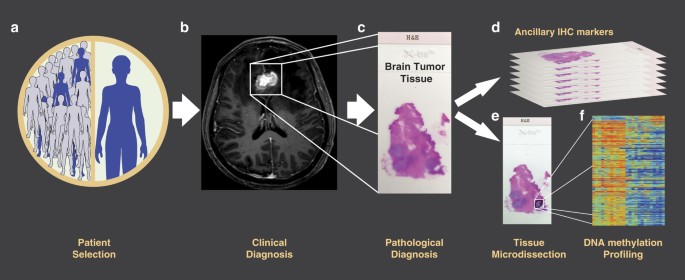
HumanMethylation 450K BeadChips (HM450K) of 96 micro-dissected BM specimens from patients with breast cancer, lung cancer, and cutaneous melanoma (Fig. 1). In addition to DNAm data, this
report provides a detailed description of the methodological approaches for patient selection, compliance matters, tissue processing and DNA preparation, data normalization, bioinformatics
analyses, and usage notes including clinical and demographic information for all patients in the study. Seven of these patients are part of a cohort study that we previously analyzed to
identify genome-wide DNAm variations during cutaneous melanoma progression to BM17–20. Therefore, the current cohort of BM DNA methylomes is composed of HM450K profiles included in two
different NCBI’s Gene Expression Omnibus (GEO) datasets (GSE108576 and GSE44661). We believe that these datasets offer a unique opportunity for the discovery of novel diagnostic and
prognostic biomarkers, while simultaneously providing insight into the underlying biology of this serious clinical complication. In this regard, we have employed these data to further
explore the utility of DNAm profiles to accurately discriminate between primary and metastatic brain tumors, identify the origin of the BM lesions, and specifically classify BCBM into
therapeutically relevant molecular subtypes21. Thus, we generated and validated a three-steps BM DNAm based classifier named "BrainMETH"21. METHODS TISSUE SPECIMEN COLLECTION A
total of 96 metastatic brain formalin-fixed paraffin-embedded (FFPE) tumor samples from 94 patients diagnosed with breast cancer BM (BCBM; n = 30), lung cancer BM (LCBM; n = 22), and
cutaneous melanoma BMs (MBM; n = 44) were included in this study. Two breast cancer patients presented synchronous or asynchronous multiple lesions. The clinical and demographic
characteristics of the patients included in the study have been summarized according to relevant information for each cancer type (Table 1). All patient-derived samples and clinical and
demographic data were collected under research protocols approved by the joint Institutional Review Board of Providence Saint John’s Health Center/John Wayne Cancer Institute, the Western
Institutional Review Board, the Institutional Review Board of Swedish Medical Center, and the Sydney Local Health District (Royal Prince Alfred Hospital Zone) Human Ethics Review Committee.
All patients signed an informed consent before joining the study. The experiments were performed in accordance with the World Medical Association Declaration of Helsinki and the National
Institutes of Health Belmont Report. Tissues were de-identified and coded according to recommendations of the Health Insurance Portability and Accountability Act (HIPAA) to ensure
confidentiality of the patients. HISTOPATHOLOGICAL CLASSIFICATION OF BRAIN METASTASIS The BCBM specimens were classified into molecular subtypes according to the expression status of the
hormone receptors (HR), i.e. estrogen receptor (ER) and progesterone receptor (PgR), and the human epidermal growth factor receptor 2 (HER2). ER and PgR were assessed by immunohistochemistry
(IHC), and HER2 by IHC and/or _in situ_ hybridization assays (ISH). FFPE tissue slides were sectioned at 4 μm, mounted onto plus-coated glass slides, and immunohistochemically stained using
a Ventana BenchMark ULTRA automated slide stainer (Roche Diagnostics, Indianapolis, IN, USA) by the Clinical Laboratory Improvement Amendments (CLIA)-certified Department of Pathology,
Providence Saint John’s Health Center, accredited by the College of American Pathologists (CAP). The antibodies used in this evaluation were the CONFIRM anti-Estrogen Receptor (SP1,
#790-4324, Ventana Medical Systems, Tucson, AZ, USA), the CONFIRM anti-Progesterone Receptor (1E2, #790-2223, Ventana Medical Systems, Tucson, AZ, USA), and the PATHWAY anti-HER-2/neu (4B5,
#790-2991, Ventana Medical Systems, Tucson, AZ, USA). The scoring criteria for these biomarkers were based on the current ASCO/CAP guidelines22,23. Briefly, ER and PgR were considered
positive if there was staining of the nucleus in at least ≥1% of tumor cells in the sample. HER2 test result was considered positive if IHC 3+ (observed in a homogeneous and contiguous
population and within >10% of the invasive tumor cells) or ISH amplified if single-probe average HER2 copy number >6.0 signals/cell or dual-probe HER2/CEP17 ratio ≥2.0. BCBM specimens
were grouped according to the expression of these routinely evaluated markers into three therapeutically relevant subgroups: a- HR positive/HER2 negative, b- HR any/HER2 positive (HER2+),
and c- HR negative/HER2 negative (aka triple-negative breast cancer; Table 1). The MBM samples were categorized according to the mutational status of _BRAF_ and _NRAS_ genes. Genomic DNA
from MBM was amplified with standardized primers specific for exon 15 of _BRAF_, and exons 1 and 2 of _NRAS_20. Polymerase chain reaction (PCR) products were purified using QIAquick® PCR
Purification Kit (#28106 Qiagen, Germany) and subsequently visualized in 2.2% agarose gel DNA cassettes for gel electrophoresis (FlashGel™ System, Lonza Inc, Rockland, ME, USA). Successfully
amplified samples were then quantified by UV absorption spectrophotometry and sequenced using an internal primer20 by Eurofins MWG Operon LCC (Eurofins Genomics LCC, Louisville, KY, USA).
Sequencing results were analyzed using Chromas Lite v2.6.5 (Technelysium Pty Ltd, Australia) and mutations in _NRAS_ and _BRAF_ genes were annotated according to the Catalogue of Somatic
Mutations in Cancer (COSMIC v86, Wellcome Sanger Institute, Cambridge, UK; http://cancer.sanger.ac.uk/cosmic)24. As the presence of _BRAF_ and _NRAS_ mutations were mutually exclusive
events, the MBM specimens were classified into 3 categories: a- BRAF mutated, b- NRAS mutated, and c- BRAF/NRAS wild-type (Table 1). Due to limited tissue availability, two specimens were
not profiled for oncogenic mutations on the _BRAF_ or _NRAS_ genes and presented in Tables 1 and 2 as not available (N/A). The LCBM were histologically classified into non-small cell lung
cancer (NSCLC) and small cell lung cancer (SCLC; Table 1). Of note, we added four BM specimens from female patients with a presumptive diagnosis of LCBM, but with inconclusive IHC analysis,
or with a previous diagnosis of both primary lung and breast cancer. In agreement with the clinical-pathological diagnosis an origin of lung cancer was confirmed by DNAm profiling21. GENOMIC
DNA EXTRACTION Representative FFPE tissue blocks from each metastatic brain lesion were selected by the respective Pathology Departments. FFPE tissue blocks were cut into 4 μm and 8 μm
serial slides. Neuropathologists reviewed 4 μm tissue slides stained with hematoxylin & eosin (H&E) for all specimens and labeled representative brain metastatic areas with an
estimated tumor purity higher than 70%. After deparaffinization, hematoxylin staining was performed in 8 μm thick serial tissue sections and needle microdissected using the labeled 4 μm
tissue slides as template. Genomic DNA (gDNA) was then isolated using ZR FFPE DNA MiniPrep (D3066; Zymo Research, Irvine, CA, USA), according to the manufacturer’s instructions. Genomic DNA
was quantified by Qubit® 3.0 Fluorometer (Q33216; Thermo Fisher Scientific, Carlsbad, CA). GENOME-WIDE DNA METHYLATION PROFILING Sodium bisulfite modification (SBM) was performed on 1 μg of
gDNA using the EZ DNA Methylation-Direct Kit (D5021, Zymo Research Irvine, CA, USA). An aliquot of SBM-DNA was analyzed by MethyLight-based quality control to test bisulfite completeness.
After correction of SBM-DNA amount, a minimum of 200 ng of SBM-DNA was whole-genome amplified and enzymatically fragmented. Finally, the fragmented SBM-DNA was hybridized into the HM450K
(Illumina Inc., San Diego, CA, USA) and scanned using the Illumina iScan microarray scanner following the manufacturer’s recommended settings (Illumina Inc., San Diego, CA, USA). DATA
PROCESSING AND ANALYSIS Data was extracted from Illumina .idat files using the Bioconductor package _minfi_25. The ‘_preprocessNoob’_ function in _minfi_ was used for normalization and
dye-bias correction as described in Triche _et al._26. DNAm levels were reported as β-values [β = intensity of the methylated allele/(intensity of the unmethylated allele + intensity of the
methylated allele)], and calculated using the signal intensity value for each CpG site. The effect of normalization on the distribution of β values across samples is shown in Fig. 2. Using
the normalized β values, we compared the genome-wide DNAm profiles for specific genomic features across the three BM groups. DNAm level of CpG sites in high-CpG density regions (known as CpG
islands; CGI) and low-CpG density regions (known as CGI shore, CGI shelves, and open sea) were also variable among the three BM groups (Fig. 3a). Additionally, DNAm levels varied among the
three BM groups for CpG sites in the promoter regions, 5’UTRs, the first exon, gene body, and intergenic regions (IGRs; Fig. 3b). Finally, to check for overall structure within our dataset,
we used the t-distributed stochastic neighbor embedding (t-SNE)27,28 method with the top 2,500 most variable HM450K probes to cluster all BM specimens. Three distinct clusters were observed
that corresponded to each of the three BM types, with MBMs showing the greatest degree of separation from BCBM and LCBM which were positioned more closely to each other (Fig. 3c). No outlier
samples were observed. CODE AVAILABILITY All analyses were performed using open source R and Bioconductor packages. Specifically, the _minfi_25 package was used to process raw array data
and perform normalizations (see “Data processing and analysis” section), summary statistics were calculated using functions in base R and the _matrixStats_29 package, density distribution
plots were generated using the _densityPlot_ function in _minfi_25, all other figures were generated using the _ggplot2_30 and _RColorBrewer_31 packages, and the t-SNE analysis was performed
using the _Rtsne_32package. No custom code was used in the processing or analysis of this data. DATA RECORDS All HM450K raw and normalized data that support the findings of this study have
been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) datasets under the series records GSE108576 (Data
Citation 1) and GSE44661 (Data Citation 2). The data is presented in a tabular format that includes the unmethylated intensity values, the methylated intensity values, the _p_-value from the
statistical evaluation of the differences between signal and noise, and the corrected β value. The DNAm data can also be accessed as raw intensity files (.idat). Additionally, the
integration of the clinical-demographic characteristics of the 96 BM specimens with the matched .idat file names and the GEO sample identifiers (GSM) is provided in Table 2 (available online
only). TECHNICAL VALIDATION To ensure that only samples with high overall quality were included in this dataset, we applied a three-step quality control pipeline: 1) We filtered samples by
probe detection _p_-value to identify samples with an elevated level of background noise. A significance level of 0.05 for the mean per sample detection _p_-value was used as a cut-off. All
96 samples included in this dataset showed mean detection _p_-values less than 0.05 (Fig. 4a). 2) We calculated the number of probes with missing β values per sample. Across all 96 samples,
the median number of probes with missing β values was 7.0 probes per sample with a range of 1 to 246 probes (Fig. 4b). Overall, the number of probes with missing β values represents a
minuscule fraction of the total number of probes present on the array and therefore is highly unlikely to have an adverse effect on downstream analysis. 3) For each probe, we calculated the
number of samples with missing β values. Notably, of the 485,577 probes included on the HM450K microarray, probe cg01550828 showed missing β values in 79 samples (Fig. 4c). Probe cg01550828
is located in the body of the ring finger protein 168 (_RNF168_) gene and is one of five probes within the _RNF168_ gene body. While cg01550828 showed missing values, none of the other four
_RNF168_ gene body probes showed any missing values across the 96 samples. USAGE NOTES To enhance the utility of this resource, we have integrated the most relevant clinical and demographic
features of the patient cohort and DNAm data for each BM specimen. In Table 2 (available online only), we included patient age at BM diagnosis, gender, primary cancer of origin, and
cancer-specific subtypes matched with GEO sample names and .idat identifiers. This information can be accessed from the respective GEO series GSE108576 (Data Citation 1) and GSE44661 (Data
Citation 2). The dataset we present here can be further analyzed to study the differential methylation profiles among the three BM groups described here and/or integrated into larger
methylation analyses using new or existing publicly available array data deposited in GEO. Data normalization and differential methylation analysis can be performed using various open source
Bioconductor packages. In particular, the ChAMP Bioconductor package provides a comprehensive analysis pipeline that utilizes many well-established methods for the normalization and
analysis of Illumina HM450K microarray data33. This package is well documented and provides a useful first pass pipeline for processing array data. ADDITIONAL INFORMATION HOW TO CITE THIS
ARTICLE: Salomon, M. P. _et al._ Brain metastasis DNA methylomes, a novel resource for the identification of biological and clinical features. _Sci. Data_. 5:180245 doi:
10.1038/sdata.2018.245 (2018). PUBLISHER’S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. REFERENCES REFERENCES
* Sperduto, P. W. et al. Summary report on the graded prognostic assessment: an accurate and facile diagnosis-specific tool to estimate survival for patients with brain metastases. _J. Clin.
Oncol._ 30, 419–425 (2012). Article Google Scholar * Soffietti, R. et al. A European Organisation for Research and Treatment of Cancer phase III trial of adjuvant whole-brain radiotherapy
versus observation in patients with one to three brain metastases from solid tumors after surgical resection or radiosurgery: quality-of-life results. _J. Clin. Oncol._ 31, 65–72 (2013).
Article Google Scholar * Lin, N. U., Bellon, J. R. & Winer, E. P. CNS metastases in breast cancer. _J. Clin. Oncol._ 22, 3608–3617 (2004). Article Google Scholar * Lin, X. &
DeAngelis, L. M. Treatment of Brain Metastases. _J. Clin. Oncol._ 33, 3475–3484 (2015). Article CAS Google Scholar * Schouten, L. J., Rutten, J., Huveneers, H. A. & Twijnstra, A.
Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. _Cancer_ 94, 2698–2705 (2002). Article Google Scholar *
Barnholtz-Sloan, J. S. et al. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. _J. Clin. Oncol._ 22,
2865–2872 (2004). Article Google Scholar * Berghoff, A. S. et al. Descriptive statistical analysis of a real life cohort of 2419 patients with brain metastases of solid cancers. _ESMO
Open_ 1, e000024 (2016). Article Google Scholar * Cagney, D. N. et al. Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: A population-based
study. _Neuro Oncol._ (2017). * Gavrilovic, I. T. & Posner, J. B. Brain metastases: epidemiology and pathophysiology. _J. Neurooncol._ 75, 5–14 (2005). Article Google Scholar *
Noushmehr, H. et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. _Cancer Cell_ 17, 510–522 (2010). Article CAS Google Scholar * Hegi,
M. E. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. _N. Engl. J. Med._ 352, 997–1003 (2005). Article CAS Google Scholar * Capper, D. et al. DNA
methylation-based classification of central nervous system tumours. _Nature_ 555, 469 (2018). Article CAS ADS Google Scholar * Moran, S. et al. Epigenetic profiling to classify cancer of
unknown primary: a multicentre, retrospective analysis. _Lancet Oncol._ 17, 1386–1395 (2016). Article Google Scholar * Ceccarelli, M. et al. Molecular Profiling Reveals Biologically
Discrete Subsets and Pathways of Progression in Diffuse Glioma. _Cell_ 164, 550–563 (2016). Article CAS Google Scholar * Sahm, F. et al. DNA methylation-based classification and grading
system for meningioma: a multicentre, retrospective analysis. _Lancet Oncol._ 18, 682–694 (2017). Article CAS Google Scholar * Brastianos, P. K. et al. Genomic Characterization of Brain
Metastases Reveals Branched Evolution and Potential Therapeutic Targets. _Cancer Discov_ 5, 1164–1177 (2015). Article CAS Google Scholar * Marzese, D. M. et al. Epigenome-wide DNA
methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. _Hum. Mol. Genet_ 23, 226–238 (2014). Article CAS
Google Scholar * Marzese, D. M., Huynh, J. L., Kawas, N. P. & Hoon, D. S. Multi-platform Genome-wide Analysis of Melanoma Progression to Brain Metastasis. _Genom. Data_ 2, 150–152
(2014). Article Google Scholar * Marzese, D. M. et al. Brain metastasis is predetermined in early stages of cutaneous melanoma by CD44v6 expression through epigenetic regulation of the
spliceosome. _Pigment Cell Melanoma Res_ 28, 82–93 (2015). Article CAS Google Scholar * Marzese, D. M. et al. DNA methylation and gene deletion analysis of brain metastases in melanoma
patients identifies mutually exclusive molecular alterations. _Neuro Oncol_ 16, 1499–1509 (2014). Article CAS Google Scholar * Orozco, J. I. J. et al. Epigenetic Profiling for the
Molecular Classification of Metastatic Brain Tumors. _Nat. Commun._ https://doi.org/10.1038/s41467-018-06715-y (2018). * Hammond, M. E. et al. American Society of Clinical Oncology/College
Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. _J. Clin. Oncol._ 28, 2784–2795 (2010). Article
Google Scholar * Wolff, A. C. et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American
Pathologists clinical practice guideline update. _J. Clin. Oncol._ 31, 3997–4013 (2013). Article Google Scholar * Forbes, S. A. et al. COSMIC: somatic cancer genetics at high-resolution.
_Nucleic Acids Res_ 45, D777–d783 (2017). Article CAS Google Scholar * Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA
methylation microarrays. _Bioinformatics_ 30, 1363–1369 (2014). Article CAS Google Scholar * Triche, T. J. Jr., Weisenberger, D. J., Van Den Berg, D., Laird, P. W. & Siegmund, K. D.
Low-level processing of Illumina Infinium DNA Methylation BeadArrays. _Nucleic Acids Res._ 41, e90 (2013). Article CAS Google Scholar * van der Maaten, L. J. P. & Hinton, G.
Visualizing Data using t-SNE. _J. Mach. Learn. Res._ 9, 2579–2605 (2008). MATH Google Scholar * van der Maaten, L. Accelerating t-SNE using Tree-Based Algorithms. _J. Mach. Learn. Res._
15, 3221–3245 (2014). MathSciNet MATH Google Scholar * Bengtsson, H. matrixStats: Functions that Apply to Rows and Columns of Matrices (and to Vectors). _R package version 0.53.1._
https://cran.r-project.org/web/packages/matrixStats/ (2018). * Wickham, H. _ggplot2: Elegant Graphics for Data Analysis_. Springer-Verlag: New York (2009). * Neuwirth, E. RColorBrewer:
ColorBrewer Palettes. _R package version 1.1-2_ https://cran.r-project.org/web/packages/RColorBrewer/index.html (2014). * Krijthe, J. H. Rtsne: T-Distributed Stochastic Neighbor Embedding
using a Barnes-Hut Implementation. _Github_ https://github.com/jkrijthe/Rtsne (2015). * Morris, T. J et al. ChAMP: 450k Chip Analysis Methylation Pipeline. _Bioinformatics_ 30, 428–430
(2014). Article CAS Google Scholar DATA CITATIONS * Salomon, M. P., Marzese, D. M., Hoon, D. S., & Orozco, J. I. _Gene Expression Omnibus_ GSE108576 (2017) * Hoon, D., & Huang, S.
_Gene Expression Omnibus_ GSE44661 (2013) Download references ACKNOWLEDGEMENTS This work was supported by the National Cancer Institute, National Institutes of Health (#R01CA167967 to DSBH)
the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to DSBH); the AVON Foundation Breast Cancer Crusade (#02-2015-061 to DMM and DSBH); the Associates for Breast and Prostate
Cancer Studies (ABCs) award (#88737700140000 to JIJO and DMM); the Fashion Footwear Association of New York (FFANY) foundation award (#88737890560000 to MPS and DSBH, #88737890550000 to
JIJO and DMM) and the John Wayne Cancer Institute Translational Research Fund (to M.P.S. and D.M.M.). AUTHOR INFORMATION Author notes * Matthew P. Salomon and Javier I. J. Orozco: These
authors contributed equally to this work. AUTHORS AND AFFILIATIONS * Department of Translational Molecular Medicine, John Wayne Cancer Institute at Providence Saint John’s Health Center,
Santa Monica, 90404, CA, USA Matthew P. Salomon, Javier I. J. Orozco, Ayla O. Manughian-Peter, Dave S. B. Hoon & Diego M. Marzese * Melanoma Institute Australia, The University of
Sydney, Camperdown, 2065, NSW, Australia James S. Wilmott & Richard A. Scolyer * Ben & Catherine Ivy Center for Advanced Brain Tumor Treatment, Swedish Neuroscience Institute,
Seattle, 98122, WA, USA Parvinder Hothi & Charles S. Cobbs * Sydney Medical School, The University of Sydney, Camperdown, 2006, NSW, Australia Richard A. Scolyer * Royal Prince Alfred
Hospital, Sydney, 2050, New South Wales, Australia Richard A. Scolyer Authors * Matthew P. Salomon View author publications You can also search for this author inPubMed Google Scholar *
Javier I. J. Orozco View author publications You can also search for this author inPubMed Google Scholar * James S. Wilmott View author publications You can also search for this author
inPubMed Google Scholar * Parvinder Hothi View author publications You can also search for this author inPubMed Google Scholar * Ayla O. Manughian-Peter View author publications You can also
search for this author inPubMed Google Scholar * Charles S. Cobbs View author publications You can also search for this author inPubMed Google Scholar * Richard A. Scolyer View author
publications You can also search for this author inPubMed Google Scholar * Dave S. B. Hoon View author publications You can also search for this author inPubMed Google Scholar * Diego M.
Marzese View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS M.P.S., J.I.J.O., D.S.B.H., and D.M.M conceptualized the study. M.P.S. processed
DNA methylation data, performed data quality controls, and compiled and transferred data to NCBI’s GEO. J.I.J.O. and A.O.M.-P. processed tissues and performed quality controls. J.S.W., P.H.,
C.S.C., and R.A.S. selected the patients, contributed with the tissue logistics, and annotation of clinical data. M.P.S. and D.M.M. constructed the figures. J.I.J.O. constructed the tables.
D.S.B.H. and D.M.M. provided general guidance and oversaw the study. M.P.S., J.I.J.O., A.O.M.-P., D.S.B.H., and D.M.M. wrote the manuscript. All authors read, edited, and approved the final
manuscript before submission. CORRESPONDING AUTHOR Correspondence to Diego M. Marzese. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ISA-TAB METADATA
ISA-TAB METADATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication
waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Salomon,
M., Orozco, J., Wilmott, J. _et al._ Brain metastasis DNA methylomes, a novel resource for the identification of biological and clinical features. _Sci Data_ 5, 180245 (2018).
https://doi.org/10.1038/sdata.2018.245 Download citation * Received: 28 June 2018 * Accepted: 03 September 2018 * Published: 06 November 2018 * DOI: https://doi.org/10.1038/sdata.2018.245
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative