A collagen glucosyltransferase drives lung adenocarcinoma progression in mice
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Cancer cells are a major source of enzymes that modify collagen to create a stiff, fibrotic tumor stroma. High collagen lysyl hydroxylase 2 (LH2) expression promotes metastasis and
is correlated with shorter survival in lung adenocarcinoma (LUAD) and other tumor types. LH2 hydroxylates lysine (Lys) residues on fibrillar collagen’s amino- and carboxy-terminal
telopeptides to create stable collagen cross-links. Here, we show that electrostatic interactions between the LH domain active site and collagen determine the unique telopeptidyl lysyl
hydroxylase (tLH) activity of LH2. However, CRISPR/Cas-9-mediated inactivation of tLH activity does not fully recapitulate the inhibitory effect of LH2 knock out on LUAD growth and
metastasis in mice, suggesting that LH2 drives LUAD progression, in part, through a tLH-independent mechanism. Protein homology modeling and biochemical studies identify an LH2 isoform
(LH2b) that has previously undetected collagen galactosylhydroxylysyl glucosyltransferase (GGT) activity determined by a loop that enhances UDP-glucose-binding in the GLT active site and is
encoded by alternatively spliced exon 13 A. CRISPR/Cas-9-mediated deletion of exon 13 A sharply reduces the growth and metastasis of LH2b-expressing LUADs in mice. These findings identify a
previously unrecognized collagen GGT activity that drives LUAD progression. SIMILAR CONTENT BEING VIEWED BY OTHERS SUPPRESSION OF PANCREATIC DUCTAL ADENOCARCINOMA GROWTH AND METASTASIS BY
FIBRILLAR COLLAGENS PRODUCED SELECTIVELY BY TUMOR CELLS Article Open access 20 April 2021 GENOME-WIDE SCREENS IDENTIFY SEL1L AS AN INTRACELLULAR RHEOSTAT CONTROLLING COLLAGEN TURNOVER
Article Open access 20 February 2024 CANCER-ASSOCIATED FIBROBLASTS REQUIRE PROLINE SYNTHESIS BY PYCR1 FOR THE DEPOSITION OF PRO-TUMORIGENIC EXTRACELLULAR MATRIX Article Open access 27 June
2022 INTRODUCTION Fibrillar collagens play a key role in maintaining tissue integrity and function1. Their structural, biochemical, and mechanical properties are regulated, in part, by
post-translational modifications of Lys residues2. Fibrillar collagens have a central triple-helical structure (“helical domain”) and amino- and carboxy-terminal non-helical “telopeptide”
domains. Both Helical Lys (hLys) and Telopeptidyl Lys (tLys) residues can be hydroxylated to form helical hydroxylysine (hHyl) and telopeptidyl hydroxylysine (tHyl). tLys and tHyl are
oxidized into allysine and hydroxyallysine, respectively, by lysyl oxidases. These aldehydes (allysine and hydroxyallysine) then undergo a series of condensation reactions with allysine,
Lys, Hyl, and histidine (His) residues to form collagen cross-links that stabilize collagen fibrils and matrices. Hydroxylation of tLys into tHyl has little impact on cross-link density but,
upon oxidation, leads to the formation of a group of structurally distinct stable collagen cross-links called Hyl aldehyde-derived collagen cross-links (HLCCs) that generate tensile
strength in load-bearing skeletal tissues like bone and cartilage2. Following oxidation and condensation, unhydroxylated tLys forms Lys aldehyde-derived collagen cross-links (LCCs) commonly
seen in soft tissues. hHyl residues in collagen can be further modified by a 2-step glycosylation (galactosylation and glucosylation) to regulate how collagens interact with collagen
receptors on cells4,5,6. Reduced Lys hydroxylation and/or glycosylation underlie inherited connective tissue disorders7,8,9,10,11, and aberrantly high HLCC production contributes to fibrotic
diseases and cancer metastasis3,12,13,14,15. Key collagen modifications are governed by lysyl hydroxylase family members (LH1-3) that have a conserved dual enzymatic domain architecture but
are functionally distinct15. While all three LH family members hydroxylate helical Lys (hLys) residues in x-Lys-glycine sequences, LH3 is reportedly unique in its ability to function in
tandem with GLT25D1/2 to convert helical Hyl residues into 1,2-glucosylgalactosyl-5-hyl through two consecutive reactions: GLT25D1/2-mediated O-linked conjugation of galactose, and
LH3-mediated conjugation of glucose to galactosyl-5-Hyl16,17,18,19. In addition, LH2 is unique in its ability to hydroxylate tLys residues, leading to the formation of stable HLCCs, as
demonstrated by evidence that inactivating mutations of the LH2-encoding gene _PLOD2_ in Bruck Syndrome are associated with HLCC deficiencies and severe skeletal abnormalities7,9. In this
study, we sought to elucidate the structural basis for LH2’s telopeptidyl LH (tLH) activity and to determine whether tLH activity underlies the pro-metastatic activity of LH2 in LUAD.
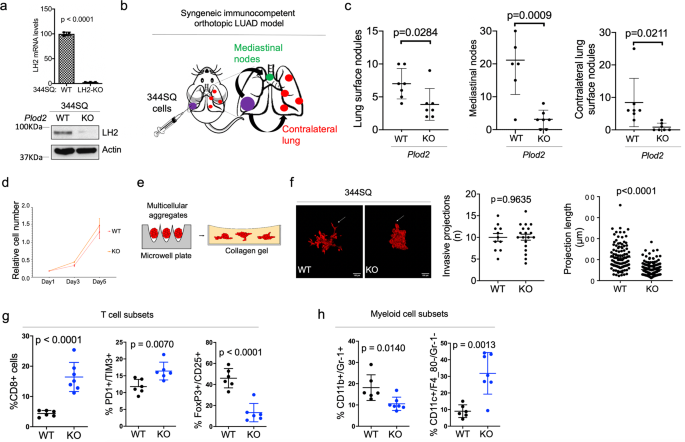
RESULTS LH2-DEPENDENT LUAD MODELS We implemented an immunocompetent LUAD model20 to identify structural features of LH2 that promote tumor growth and metastasis. Relative to parental
_K-ras/Tp53_-mutant 344SQ LUAD cells, 344SQ cells subjected to CRISPR/Cas-9-mediated _Plod2_ knockout (KO) generated orthotopic LUADs that were smaller and less metastatic to mediastinal
lymph nodes and the contralateral lung (Fig. 1a-c, Supplementary Fig. 1). Compared to parental cells, _Plod2_ KO 344SQ cells demonstrated no loss of proliferative activity in monolayer
culture (Fig. 1d) but generated multicellular aggregates that were less invasive in collagen gels (Fig. 1e, f), which is in line with evidence that LH2 promotes LUAD invasive activity3.
Enhanced intra-tumoral fibrosis is associated with reduced T cell infiltration, M2 macrophage polarization, and increased recruitment of regulatory T cells and myeloid-derived suppressor
cells that inhibit CD8+ T cell immunity21. Immune cells express collagen receptors that have immunosuppressive functions22. To determine whether LH2 influences intra-tumoral immunity, we
quantified immune cell subsets in subcutaneous tumors generated by _Plod2_ KO or parental 344SQ cells and identified alterations in T cell and myeloid cell subsets that were consistent with
an anti-tumor response in _Plod2_ KO tumors (Fig. 1g, h). Thus, LH2 influences collagen’s immunosuppressive functions. ELECTROSTATIC INTERACTIONS SPECIFY TLH ACTIVITY Given that the LH
domain active site is highly conserved across LH family members23, we reasoned that residues extrinsic to LH2’s active site determine tLH activity. To identify the domain in which those
residues reside, we performed rescue experiments on LH2-deficient MC3T3-E1 (MC) osteoblasts and found that residues required for HLCC reconstitution reside in LH2’s LH domain (Fig. 2a-g,
Supplementary Fig. 2). LH2 homology modeling based on a mimiviral tLH domain crystal structure (Fig. 2h) identified 2 basic amino acid residues (R680 and R682) adjacent to the LH2 active
site that are dramatically different in charge and hydrophobicity from the corresponding amino acids in LH1 and LH3 (Fig. 2h, i). Furthermore, highly acidic aspartate and glutamate residues
are positioned adjacent to tLys residues on fibrillar collagens (Supplementary Fig. 3). Replacing R680 and R682 on LH2 with the corresponding residues on LH1 (LH2-EP) ablated tLH activity
(Fig. 2j), whereas activity on type I collagen, which contains hLys residues, was relatively preserved (Fig. 2k). Conversely, replacing collagen telopeptide’s two acidic residues with
alanine reduced the activity of wild-type, but not LH1-mimic, LH2 (Fig. 2l, m). These data suggest that LH2’s unique tLH activity is determined by electrostatic interactions between LH2 and
collagen telopeptides. TLH INACTIVATION DOES NOT RECAPITULATE THE EFFECT OF _PLOD2_ KO Based on the temporal relationship between stable collagen cross-link accumulations and enhanced tumor
cell invasion and metastasis3,13,24,25,26, we postulated that tLH activity accelerates LUAD progression. To test this hypothesis, we ablated tLH activity in 344SQ cells by introducing
mutations that reduce Fe2+-binding in the active site or disrupt LH2 dimer assemblies (Supplementary Fig. 4) without altering LH2 protein levels in 344SQ cells (Supplementary Fig. 5). These
mutations reduced orthotopic LUAD metastatic capacity but not size (Fig. 2n, o) and did not fully recapitulate the effect of _Plod2_ KO (Fig. 1a-c), suggesting that LH2 drives LUAD
progression through tLH-dependent and -independent mechanisms. LH1 AND LH2 HAVE COLLAGEN GGT ACTIVITY With respect to potential tLH-independent mechanisms of action, we reasoned that the GLT
domain of LH2 may have enzymatic activity that escaped previous detection27. By sequence alignment, the LH3 GLT domain has 60 and 57% sequence identity to the corresponding domains of LH1
and LH2, respectively, and the DXXD motif28 is strictly conserved in LH1 but not LH2; D112 and Y114 in LH3 are replaced with glutamate and phenylalanine, respectively, in LH2 to preserve
charge and hydrophobicity (Fig. 3a, Supplementary Fig. 6). To determine whether LH1 and LH2 have galactosylhydroxylysyl glucosyltransferase (GGT) activities, we developed a luciferase assay
that detects UDP release following reaction of recombinant LH proteins with UDP-glucose and a synthetic amino acid substrate (galactosyl-Hyl) or deglucosylated type IV collagen.
Deglucosylation of type IV collagen was achieved by treatment with a collagen glucosidase, protein-glucosylgalactosylhydroxylysine glucosidase (PGGHG) (Supplementary Fig. 7)29. Under these
conditions, all LH family members had detectable GGT activities that were abolished by mutation of Mn2+-binding residues or omission of PGGHG pretreatment (Fig. 3b-h). Because key residues
in LH3’s GLT active site are only partially conserved in LH2, we speculated that LH2’s GGT activity has a distinct structural basis. LH2 is alternatively spliced into isoforms that do (LH2b)
or do not (LH2a) include exon 13A, which encodes 21 amino acids that are reported to regulate tLH activity30,31. However, LH2a and LH2b similarly rescued collagen crosslinking defects in
LH2-deficient MC cells (Fig. 3I, j, Supplementary Figs. 8 and 9) and had comparable tLH and hLH activities in enzymatic assays (Fig. 3k, Supplementary Fig. 10). In contrast, GGT activity was
sharply higher in LH2b (Fig. 3l, m). To determine how alternative splicing regulates LH2’s GGT activity, we modeled LH2b using the recently determined full-length LH3 structure28 and found
that exon 13A adopts a loop conformation that is positioned between α10 and β14 of an accessory domain with a Rossmann fold in close proximity to the GLT active site (Fig. 3n), which led us
to speculate that the loop influences substrate binding affinity. By microscale thermophoresis, binding affinity to fluorescein-conjugated UDP-glucose was higher for LH2b than LH2a (Fig.
3o), and the isoforms demonstrated distinct binding modes (Supplementary Fig. 11a), suggesting that the exon 13A-encoded loop may enhance collagen GGT activity by functioning as a GLT active
site cap. Unlabeled UDP-glucose competed with fluorescein-conjugated UDP-glucose for binding to LH2b with an IC50 of 30 µM (Supplementary Fig. 11b), suggesting that fluorescein is not
involved in binding. Thus, LH2b is a collagen GLT that is regulated by cooperative interactions between tandem Rossmann domains. LH2B DRIVES LUAD GROWTH AND METASTASIS
Glucosylgalactosyl-dihydroxylysinonorleucine (GG-DHLNL) levels were higher in human LUAD than they were in adjacent normal lung tissues (Fig. 4a), warranting studies to determine how
collagen glycosylation is regulated in LUAD and its role in LUAD progression. In The Cancer Genome Atlas lung cancer cohorts, which are mostly early-stage tumors, LH2a is the predominant
isoform, whereas normal lung tissues have similarly low levels of the 2 isoforms (Fig. 4b-d). In contrast, LH2b levels were equal to or higher than LH2a levels in human LUAD cell lines and
highly, but not poorly, metastatic LUAD cell lines derived from _K-ras/Tp53_-mutant mice (Fig. 4e, Supplementary Fig. 12-14)20. Thus, the predominant LH2 isoform switches from LH2a to LH2b
during LUAD progression. Splicing factors that drive exon 13 A inclusion and thereby increase the relative levels of LH2b have been identified32,33,34. One of them, FOX2, is of particular
interest because it regulates alternative splicing driven by epithelial-to-mesenchymal transition (EMT)35,36, which initiates metastasis in _K-ras/Tp53_-mutant LUAD models20,37,38,39. Small
interfering RNA-mediated depletion of FOX2 in 344SQ cells decreased LH2b levels (Fig. 4f, g, Supplementary Fig. 15), indicating that FOX2 promotes exon 13 A inclusion in 344SQ cells. We
subjected 344SQ cells to CRISPR/Cas-9-mediated deletion of exon 13 A to examine the consequences of LH2b loss on LUAD progression. Relative to parental cells, 344SQ_Δexon 13 A cells had no
detectable change in total LH2 protein levels (Supplementary Fig. 5) but demonstrated reduced LH2b and increased LH2a mRNA levels (Supplementary Fig. 16). Orthotopic LUADs and flank tumors
generated by 344SQ_Δexon 13A cells in syngeneic, immunocompetent mice were smaller and less metastatic than those generated by parental 344SQ cells (Fig. 4h, i). Multicellular aggregates
generated by 344SQ_Δexon 13 A cells were less invasive than aggregates generated by parental or tLH-deficient (L735D) 344SQ cells (Fig. 4j, k). Orthotopic LUADs in nu/nu mice injected with
an exon 13A-deficient H358 human LUAD cell line were less metastatic than those generated by parental H358 cells (Fig. 4l), but this difference did not reach statistical significance,
potentially owing to low baseline metastatic activity of the orthotopic LUADs or the absence of an intact immune system. The contribution of intratumoral immunity was difficult to ascertain
given that the subcutaneous tumors generated by 344SQ_Δexon 13A cells were too small for flow cytometric analysis. Thus, LH2b’s GGT activity drives LUAD growth and metastasis. DISCUSSION The
presence of a fibrotic tumor stroma is correlated with hypoxia, immunosuppression, treatment resistance, and metastasis14. These features result in part from an accumulation of stable
collagen cross-links3,24]. Cancer cells direct the formation of stable collagen cross-links by producing enzymes that hydroxylate (e.g., LH2) or oxidatively deaminate (e.g., lysyl oxidases)
Lys residues on collagen3,24. Here, we abolished tLH activity by disrupting either Fe2+-binding in the active site or LH2 dimer assembly formation and found that LUAD progression was
minimally impaired. Homology modeling and biochemical studies identified collagen GGT activity that results from an alternatively spliced exon in LH2. These findings identify a previously
unrecognized collagen GGT activity that may enable cancer growth and metastasis. Collagen glycosylation has a profound impact on collagen fibrillogenesis, cross-link maturation, collagen
stability, matrix mineralization, axonal guidance, and platelet activation40,41,42,43,44. Glycosylation influences the ability of collagens to function as ligands for receptors (e.g., DDRs,
integrins, CD44) that direct cellular functions during embryonic development and tissue repair and are aberrantly expressed in numerous pathologic conditions, including cancer4,5,6. Based on
our finding that LH1 and LH2 have collagen GGT activity, we conclude that all LH family members are bifunctional enzymes that function within an integrated collagen regulatory network, and
future studies should readdress polymorphisms and inherited mutations in GLT domains of the _PLOD_ gene family that were previously thought to be non-contributory in individuals with
connective tissue disorders45,46. Moreover, our finding that Hyl glucosylation is a pro-tumorigenic collagen modification opens new avenues of research into how collagen glucosylation
influences pro-metastatic processes in the tumor microenvironment. It remains unclear whether LH2- and LH3-catalyzed GGT activities play distinct or overlapping biochemical and biological
roles18. LH2 and LH3 may glucosylate distinct G-Hyl residues on collagen and/or modify different collagen types. The finding that LH2’s GGT activity is determined by an alternatively spliced
exon raises the possibility that its dual enzymatic functions are coordinately regulated. Such coordinated activities could profoundly influence angiogenesis and other LH family-dependent
processes in the tumor microenvironment as recently reported47. Although further studies are necessary to dissect the molecular events that link collagen GGT activity to cancer progression,
our data raise the tantalizing possibility that drugs that target LH2’s GLT active site could be useful for the treatment of LUAD and other LH2-dependent cancer types. Finally, by performing
domain swapping and rescue experiments on LH2-deficient osteoblasts, we showed that LH2’s LH domain determines its tLH activity. Site-directed mutagenesis and enzymatic activity assays
identified residues that are critical for LH2’s tLH activity. These results indirectly suggest that a unique arginine cluster near LH2 active site may directly engage acidic residues in
collagen telopeptides to facilitate substrate recognition. However, protein crystallographic studies will be required to directly substantiate this possibility. METHODS PLASMIDS Full-length
murine PGGHG was cloned into a modified pET-28b (Novagen) vector using NheI and NotI cloning sites with standard PCR-based methods. This modified pET-28b vector has NheI inserted in the
linker between His6 and Thrombin recognition site, which changes the amino acid linker from GS to AS. For recombinant protein production in Chinese hamster ovary (CHO) cells, truncated human
LH1 (aa22-727) and LH3 (aa32-738) were cloned into BamH1 and Not1 sites of pSGHP1, which was a generous gift from Dr. Craig W. Vander Kooi (University of Kentucky). Truncated human LH2
(residues 33-737 for LH2a and residues 33–758 for LH2b) were cloned into XbaI and Not1 sites of pSGHP1. Point mutant constructs were generated using QuickChange Lightning Site-Directed
Mutagenesis Kit (Agilent). For ectopic expression in MC cells, murine LH1, LH2a, LH2b and mutant were cloned into XbaI and NotI cloning sites of pEF-bsr with standard PCR-based methods48.
The identities of all constructs used in this study were confirmed by sequencing. Primers used for cloning and mutagenesis are listed (Supplementary Table 1). CRISPR/CAS-9 _PLOD2_ EDITING
344SQ and H358 cells were cultured in Roswell Park Memorial Institute 1640 supplemented with 10% FBS (complete media) in a humidified atmosphere with 5% CO2 at 37 °C. To generate _Plod2_ KO
344SQ cells, we re-constructed the Cas9-2A-GFP vector (Sigma) to express guide RNAs that flank exon1 of _Plod2_ under the U6 promoter (Supplementary Table 2). 344SQ cells were transiently
transfected with vector using lipofectamine 2000 transfection reagent (Thermo Fisher). Three days later, the top 5% of GFP+ cells were isolated by flow sorting and plated as single cells by
limiting dilution method. To screen the clones, genomic DNA was extracted from the cells using QuickExtract™ DNA Extraction Solution (Epibio, Inc) and amplified by PCR with PCR primers
flanking the deleted region. The deletion was confirmed by quantitative RT-PCR analysis of RNA and Western blot analysis of cell lysates. For D689A and L735D knock-ins, cells were
electroporated with 7 µg all-in-one Cas9/gRNA vector and 0.3 nmol ssODN donor template (Supplementary Table 2). For exon 13 A deletion, cells were electroporated with 10 µg all-in-one
Cas9/gRNA vectors. Electroporated cells were allowed to recover for 48 h before being sorted for GFP+ cells and were then plated by limiting dilution at <1 cell per well into 96-well
plates in complete medium. Once clones grew to acceptable sizes, they were expanded into 24-well plates and then processed to extract mRNA and genomic DNA for PCR amplification. PCR products
were then digested using the SalI enzyme to identify D689A Knock-in clones or digested using the SmaI enzyme to identify L735D Knock-in clones or subjected to gel electrophoresis to
identify exon 13A-deleted clones. D689A and L735D knock-in clones were further confirmed by Sanger sequencing. TUMOR MODELS Immunocompetent 129/Sv mice syngeneic to 344SQ cells were bred
in-house and randomized to balance the cohorts based on age. Nude mice were purchased from The University of Texas MD Anderson Cancer Center ERO department. Mice were placed under general
anesthesia (ketamine/xylazine 50 mg kg−1 and 5 mg kg−1, respectively, delivered by intraperitoneal injection) and an incision was made on the thorax under sterile conditions to expose the
left lung. Tumor cells (106) were injected directly into the left lung in 50 μl sterile PBS. The incision was closed using staples. The mice were humanely killed after 8 days (344SQ exon
13A-deleted mutants) or 7 days (344SQ LH-inactive mutants) or 7 weeks (H358 exon 13A-deleted mutants) post injection. Metastatic tumors visible on the surface of mediastinal lymph nodes or
the right lung were manually counted. Investigators were blinded to the cohorts at the time of assessment of metastatic tumor numbers. Mice were excluded from the analysis if they died at
the time of tumor cell injection due to hemorrhage or pneumothorax. For flow cytometry analysis, syngeneic 129/Sv mice received subcutaneous injections of parental or CRISPR/Cas-9-edited
344SQ cells (1 × 106 per mouse) in the right flank. The mice were monitored for tumor growth and euthanized 3 weeks after the injection or at the first sign of morbidity. They were
necropsied to isolate the primary tumors for flow cytometric analysis. Measurements were taken from a single tumor generated in each mouse (_n_ = 8–10 mice per cohort). FLOW CYTOMETRY Tumors
were dissociated using the MACS (Miltenyi) mouse tumor dissociation kit. Mechanical dissociation using a gentleMACS Octo dissociator (Miltenyi) was performed followed with enzymatic
digestion with collagenase I (0.05% w/v, Sigma), DNase type IV (30 U/ml, Sigma), and hyaluronidase type V (0.01% w/v, Sigma) for 40 min with rocking at 37 °C. Tumor samples were mechanically
dissociated again and passed through a 70 μm filter before being stained with fluorochrome-conjugated antibodies in FACS buffer. RBC lysis (Biolegend) was performed on both single cell
tumor and splenocytes samples following manufacturer recommendation. Cells were stained for surface markers using fluorochrome-conjugated anti-mouse antibodies (CD45, CD3, CD8, CD4) for 1 h
at room temperature. Ghost aqua BV510 (Tonobo) was used to stain dead cells. Cells were fixed using 1% PFA at room temperature for 15 min, then washed twice with perm/wash buffer
(Biolegend). Cells were stained with primary antibodies at room temperature for 1 h. Cells were filtered and analyzed on BD LSR Fortessa (BD Biosciences) and analyzed using FlowJo software
(v.10.5.3; Tree Star). For single color compensation, ultracomp eBeads compensation beads (Thermo Fisher) were used and stained with a single fluorescent-conjugated antibody according to
manufacturer’s instructions. Compensation was calculated automatically using BD FACSDiva 8.0.1. Gating schemes utilized for flow cytometry analysis of T cell subsets and myeloid cells are
shown in Supplementary Figs. 17 and 18. The antibodies used (clone, dilution, company, catalogue #) are as follows: CD8 PE-Cy7 (53-6.7) 1/800FCBioLegend/100721, CD3 PE-594 (17A2) 1/100
FCBioLegend/100246, CD4 APC-Cy7 (RM4-5) 1/100 FCBioLegend/100526, FoxP3 PerCp-Cy5.5 (FJK-16s) 1/100 FCeBioscience/45-5773-82, CD45 Pacific Blue (30-F11) 1/100 FCBioLegend/103126, CD25 BUV395
(PC61) 1/100 FCBD Biosciences/564022, CD11c BV786 (N418) 1/100 FCBioLegend/117335, GR1 BV711 (RB6-8C5) 1/100 FCBioLegend/108443, TIM3 APC (B8.2c12) 1/100 FCBioLegend/134007, PD1 BV605 (29
F.1A12) 1/100 FCBioLegend/135220, F4/80 APC (BM8.1) 1/100 FCTonbo/20-4801-U100, CD11b BV650 (M1170) 1/100 FCBioLegend/101239, Ghost aqua BV510 1/50 FCTonbo. CELL PROLIFERATION Viable cell
densities were quantified in subconfluent culture conditions using water-soluble tetrazolium salt 1 (WST1) reagent as suggested by manufacturer’s instructions (Takara). Measurements were
taken from distinct samples. Mean values determined from replicate (_n_ ≥ 3) biological samples. SIRNA KNOCKDOWN FOX2 siRNAs were purchased from Sigma (SASI_Mm02_00305828 and
SASI_Mm02_00305829). 344SQ cells were transfected with 100 nM control siRNAs or siRNAs against FOX2. Total RNAs were extracted from the transfected cells 48 h later and cDNA were generated
from the RNA samples using qScript cDNA SuperMix (QuantaBio). The expression levels of PLOD2 isoforms were determined using Real-Time PCR. MULTICELLULAR AGGREGATES Multicellular aggregates
were created in a 24-well plate containing 1700 laser-ablated microwells per well as described49. The microwells were passivated with 0.05% pluronic acid for 1 h prior to seeding the cells.
85,000 LUAD cells were seeded per well and cultured for 48 h to generate aggregates, each containing 50 LUAD cells. To examine invasive projection formation on multicellular aggregates in
collagen gels, multicellular aggregates were mixed with rat tail-derived type I collagen to generate collagen gels with defined concentrations (2 mg ml−1), volumes (200 µl per gel), and
aggregate densities (35 aggregates per gel). The gels were allowed to polymerize upside-down on a glass-bottom 35 mm dish at 37 °C for 30 min. The aggregates were cultured for up to 3 days,
fixed and stained with phalloidin for visualization, and imaged with a NikonA1 confocal microscope, 10× objective. Invasive projections were defined as at least one visible LUAD cell
protruding out of the aggregate. The length of the projections was manually quantified using the hand free tool to follow the projection shape (ImageJ). Measurements were taken from distinct
samples. Mean values determined from replicate (_n_ ≥ 3) biological samples. PROTEIN EXPRESSION AND PURIFICATION PGGHG was expressed in _E. coli_ strain Rosetta (DE3). Cells expressing
PGGHG were induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 16 h at 16 °C. Cells were collected, pelleted and then resuspended in binding buffer (20 mM Tris, pH 8.0, 200 mM
NaCl and 15 mM imidazole). The cells were lysed by sonication and then centrifuged at 23,000 _g_ for 15 min. The recombinant PGGHG proteins (wild type or D300E inactive mutant) were
purified with immobilized metal affinity chromatography. Human LH1-3 recombinant proteins were purified from CHO cell–derived conditioned medium samples as described previously50. In brief,
LH1-3 recombinant proteins were transiently transfected in new Gibco™ ExpiCHO™ cells (Thermo Fisher Scientific, Waltham, MA) with polyethylenimine and expressed as a secreted protein with
N-terminal His8 and human growth hormone (hGH) tags via large-scale suspension culture. The LH1-3–containing conditioned medium samples were harvested by centrifugation at 7000 _rpm_ for 10
min, filtered through 0.22 μm EMD Millipore Stericup™ Sterile Vacuum Filter Units (EMD Millipore, Billerica, MA), concentrated to 100 mL, and buffer-exchanged into Nickel-binding buffer (20
mM Tris, 200 mM NaCl, 15 mM imidazole, pH 8.0) using the Centramate™ & Centramate PE Lab Tangential Flow System (Pall Life Sciences, Ann Arbor, MI). The recombinant LH proteins were then
purified with tandem immobilized metal affinity chromatography and anion exchange chromatography. STRUCTURE MODELLING Human LH2 LH domain and LH2b full length structure homologies were
generated by the SWISS-MODEL (Swiss Institute of Bioinformatics, Biozentrum, University of Basel, Switzerland) homology server51. PDB entry 6AX7 and 6FXT were utilized as templates to model
LH2 LH domain and LH2b full length, respectively. LH ENZYMATIC ACTIVITY ASSAY LH enzymatic activity was measured using a luciferase-based assay as described50. In brief, the assay was
performed in LH reaction buffer (50 mM HEPES buffer pH 7.4, 150 mM NaCl) at 37 °C for 1 h with 1 μM LH enzymes, 10 μM FeSO4, 100 μM 2-OG, 500 μM ascorbate, 1 mM dithiothreitol, 0.01% triton
x-100, and 1 mM wild type (LSYGYDEKSTGGISVP(GPO)8) or mutant (LSYGYAAKSTGGISVP(GPO)8) collagen telopeptide mimics or 4 μM bovine skin collagen substrate containing no telopeptides (Bovine
PureCol®, Advanced BioMatrix). Collagen telopeptide mimics were dissolved in reaction buffer and incubated overnight at 4 °C to facilitate the formation of trimers, which was confirmed by
circular dichroism spectroscopy (Supplementary Fig. 19). Except for LH recombinant protein and bovine skin collagen, all reagents were prepared immediately before use. All these reagents
were dissolved in reaction buffer except for FeSO4 and collagen, which was prepared in 10 mM HCl, and the pH of the reaction mixture was checked with pH papers to ensure that HCl did not
change the overall sample pH. Bovine skin collagen was denatured by heating at 95 °C for 5 min and then chilled immediately on ice before use. LH activity was measured by detecting succinate
production with an adenosine triphosphate–based luciferase assay (Succinate-Glo™ JmjC Demethylase/Hydroxylase Assay, Promega, Madison, WI) according to manufacturers’ instructions.
Experiments were performed in triplicate from distinct samples, and an unpaired _t_-test was used to compare the enzymatic activity of different samples. CIRCULAR DICHROISM SPECTROSCOPY LH2
recombinant proteins or synthetic telopeptide mimics were analyzed in 0.01 M sodium phosphate and 150 mM NaCl (pH 7.4) at a concentration of 0.5 mg ml−1. Circular dichroism spectra were
measured using a J-810 spectropolarimeter (Jasco, Easton, MD) with a 2 mm path length quartz cuvette. All measurements were performed at 20 °C and three scans averaged for each spectrum. A
blank spectrum of phosphate-buffered saline was collected in the same manner and used for background subtraction. Results represent the mean values from triplicate technical repeats in a
single experiment. Each protein was analyzed twice. TYPE IV COLLAGEN DEGLUCOSYLATION Human type IV collagen (MilliporeSigma, St. Louis, MO) was deglucosylated in deglucosylation reaction
buffer (50 mM acetate buffer pH5.3, 150 mM NaCl) at 37 °C for 4 h with 100 ug of PGGHG enzymes and 2500 µg type IV collagen. Type IV collagen treated with inactive PGGHG D300E mutant served
as a negative control. After incubation, the reaction was stopped by incubating at 98 °C for 3 min. The deglucosylated type IV collagen (dgCol4) production was indirectly detected by
measuring glucose release using Glucose Colorimetric/Fluorometric Assay Kit (MilliporeSigma, St. Louis, MO) according to manufacturers’ instructions. Experiments were performed in duplicate
from distinct samples, and an unpaired _t_-test was used to compare the enzymatic activity of different samples. GGT ENZYMATIC ACTIVITY ASSAY GGT activity was measured in reaction buffer
(100 mM HEPES buffer pH 8.0, 150 mM NaCl) at 37 °C for 1 h with 1 μM LH enzymes, 20 μM MnCl2, 100 μM UDP-glucose (MilliporeSigma, St. Louis, MO), 1 mM dithiothreitol, 0.02% bovine serum
albumin, and 1 mM galactosyl hydroxylysine (Cayman Chemical, Ann Arbor, MI) or 2 μM dgCol4. GGT activity was measured by detecting UDP production with an ATP-based luciferase assay (UDP-Glo™
Glycosyltransferase Assay, Promega, Madison, WI) according to manufacturers’ instructions. Experiments were performed in triplicate from distinct samples, and an unpaired _t_-test was used
to compare the enzymatic activity of different samples. WESTERN BLOT Cells were washed with PBS and lysed with cell lysis buffer (Cell Signaling Technology, Danvers, MA) to extract total
proteins. Cell lysates were separated by SDS-PAGE, transferred onto nitrocellulose transfer membrane using Trans-Blot Turbo Transfer System (Bio-Rad), and then incubated with primary
antibodies and horseradish peroxidase-conjugated secondary antibodies (Proteintech, Sigma and GE Healthcare). Protein bands were visualized using Pierce ECL Western blotting substrate
(Thermo Fisher Scientific). Experiments were performed in triplicate from distinct samples. Results are representative of replicate experiments. MICROSCALE THERMOPHORESIS
Glucose-UDP-(PEG)6-Fluorescein Conjugate (10 μl at 50 nM, Sigma) was mixed with equal volume of serially diluted unlabeled LH2 proteins in 20 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM Mn2+, 0.05%
Tween-20. After incubation at 25 °C for 15 min, the samples were loaded into silica capillaries (Nanotemper Technologies). For the competition assay, fixed concentrations of fluorescein
conjugated UDP-Glucose (50 nM) and LH2b (20 µM) were titrated with different concentrations of unlabeled UDP-Glucose. Measurements were performed at 20 °C using Monolith NT.115 (Nanotemper
Technologies). Data were analyzed (Nanotemper Analysis software. v.1.2.101) to fit Kd according to the law of mass action and to determine IC50. The experiment was repeated once. Results
represent the mean values from duplicate biological samples. COLLAGEN CROSS-LINK ANALYSES For collagen cross-link analysis, MC cells were cultured for 2 weeks as described. The cell/matrix
layer was washed with cold PBS, scraped, collected and pelleted by centrifugation at 10,000 _rpm_ for 30 min. The pellets were washed with cold PBS and distilled water, lyophilized, weighed
and aliquoted. Aliquots were reduced with standardized NaB3H4, acid hydrolyzed and subjected to amino acid and cross-link analyses as reported52. The reducible cross-links, dehydro
(deH)-dihydroxylysinonorleucine/its ketoamine, deH-hydroxylysinonorleucine/its ketoamine and deH-histidinohydroxymerodesmosine (for cross-link chemistry, see2) were analyzed as their reduced
forms, i.e., DHLNL, HLNL and HHMD, respectively, and the mature trivalent cross-links pyridinoline and deoxypyridinoline were simultaneously quantified by their fluorescence. All
cross-links were quantified as mol/mol of collagen based on the value of 300 residues of hydroxyproline per collagen molecule. The Hyl content in collagen was calculated as Hyl/Hyp X 300.
Experiments were performed in triplicate from distinct samples. Mean values determined from replicate (_n_ = 3) biological samples. Because the O-glycosidic linkage of the carbohydrate
remains intact in base hydrolysis, the glycosylated immature bifunctional cross-link (GG-DHLNL) was analyzed by subjecting human LUAD to base hydrolysis with 2 N NaOH and analyzed as
described previously18. STATISTICS AND REPRODUCIBILITY Measurements were taken from replicate biological samples. Results are representative of replicate experiments. Mean values were
determined from replicate (_n_ ≥ 3) biological samples. Statistical significance was determined using 2-tailed Student’s _t_ test. Whenever possible, investigators were blinded to the
treatment groups at the time of assessment. STUDY APPROVAL All mouse studies were approved by the Institutional Animal Care and Use Committee at The University of Texas MD Anderson Cancer
Center. The use of lung tissues quantification of collagen cross-links in this study was performed under Institutional Review Board–approved protocol IRB(2)0910-01565x at Houston Methodist
Research Institute, and written informed consent was obtained from participants or their guardians. REPORTING SUMMARY Further information on research design is available in the Nature
Research Reporting Summary linked to this article. DATA AVAILABILITY Source data underlying plots shown in figures are provided in Supplementary Data 1. All other data, if any, will be
available upon reasonable request. REFERENCES * Guimarães C. F., Gasperini L., Marques A. P., Reis R. L. The stiffness of living tissues and its implications for tissue engineering. _Nat.
Rev. Mater._ 5, 351–370 (2020). * Yamauchi, M. & Sricholpech, M. Lysine post-translational modifications of collagen. _Essays Biochem_. 52, 113–133 (2012). Article CAS PubMed PubMed
Central Google Scholar * Chen, Y. et al. Lysyl hydroxylase 2 induces a collagen cross-link switch in tumor stroma. _J. Clin. Investig._ 125, 1147–1162 (2015). Article PubMed PubMed
Central Google Scholar * Vogel, W., Gish, G. D., Alves, F. & Pawson, T. The discoidin domain receptor tyrosine kinases are activated by collagen. _Mol. Cell_ 1, 13–23 (1997). Article
CAS PubMed Google Scholar * Stawikowski, M. J., Aukszi, B., Stawikowska, R., Cudic, M. & Fields, G. B. Glycosylation modulates melanoma cell alpha2beta1 and alpha3beta1 integrin
interactions with type IV collagen. _J. Biol. Chem._ 289, 21591–21604 (2014). Article PubMed PubMed Central CAS Google Scholar * Lauer-Fields, J. L., Malkar, N. B., Richet, G., Drauz,
K. & Fields, G. B. Melanoma cell CD44 interaction with the alpha 1(IV)1263-1277 region from basement membrane collagen is modulated by ligand glycosylation. _J. Biol. Chem._ 278,
14321–14330 (2003). Article CAS PubMed Google Scholar * Scietti L., Campioni M., Forneris F. SiMPLOD, a Structure-Integrated Database of Collagen Lysyl Hydroxylase (LH/PLOD) Enzyme
Variants. _J. Bone Miner. Res._ 34, 1376–1382 (2019). * Hyland, J. et al. A homozygous stop codon in the lysyl hydroxylase gene in two siblings with Ehlers-Danlos syndrome type VI. _Nat.
Genet._ 2, 228–231 (1992). Article CAS PubMed Google Scholar * Ha-Vinh, R. et al. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures
of the large joints) caused by a recessive mutation in PLOD2. _Am. J. Med. Genet. A_ 131, 115–120 (2004). Article PubMed Google Scholar * Vahidnezhad, H. et al. Mutations in PLOD3,
encoding lysyl hydroxylase 3, cause a complex connective tissue disorder including recessive dystrophic epidermolysis bullosa-like blistering phenotype with abnormal anchoring fibrils and
type VII collagen deficiency. _Matrix Biol._ 81, 91–106 (2019). Article CAS PubMed Google Scholar * Salo, A. M. et al. A connective tissue disorder caused by mutations of the lysyl
hydroxylase 3 gene. _Am. J. Hum. Genet_ 83, 495–503 (2008). Article CAS PubMed PubMed Central Google Scholar * van der Slot, A. J. et al. Increased formation of pyridinoline cross-links
due to higher telopeptide lysyl hydroxylase levels is a general fibrotic phenomenon. _Matrix Biol._ 23, 251–257 (2004). Article PubMed CAS Google Scholar * Eisinger-Mathason, T. S. et
al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. _Cancer Disco._ 3, 1190–1205 (2013). Article CAS Google Scholar * Yamauchi, M., Barker, T. H.,
Gibbons, D. L. & Kurie, J. M. The fibrotic tumor stroma. _J. Clin. Investig._ 128, 16–25 (2018). Article PubMed PubMed Central Google Scholar * Piersma B., Bank R. A. Collagen
cross-linking mediated by lysyl hydroxylase 2: an enzymatic battlefield to combat fibrosis. _Essays Biochem_. 63, 377–387 (2019). * Myllyla, R. et al. Expanding the lysyl hydroxylase
toolbox: new insights into the localization and activities of lysyl hydroxylase 3 (LH3). _J. Cell Physiol._ 212, 323–329 (2007). Article CAS PubMed Google Scholar * Schegg, B.,
Hulsmeier, A. J., Rutschmann, C., Maag, C. & Hennet, T. Core glycosylation of collagen is initiated by two beta(1-O)galactosyltransferases. _Mol. Cell Biol._ 29, 943–952 (2009). Article
CAS PubMed Google Scholar * Sricholpech, M. et al. Lysyl hydroxylase 3 glucosylates galactosylhydroxylysine residues in type I collagen in osteoblast culture. _J. Biol. Chem._ 286,
8846–8856 (2011). Article CAS PubMed PubMed Central Google Scholar * Terajima, M. et al. Role of Glycosyltransferase 25 Domain 1 in Type I Collagen Glycosylation and Molecular
Phenotypes. _Biochemistry_ 58, 5040–5051 (2019). Article CAS PubMed Google Scholar * Gibbons, D. L. et al. Contextual extracellular cues promote tumor cell EMT and metastasis by
regulating miR-200 family expression. _Genes Dev._ 23, 2140–2151 (2009). Article CAS PubMed PubMed Central Google Scholar * Jiang, H., Hegde, S. & DeNardo, D. G. Tumor-associated
fibrosis as a regulator of tumor immunity and response to immunotherapy. _Cancer Immunol. Immunother._ 66, 1037–1048 (2017). Article CAS PubMed PubMed Central Google Scholar * Meyaard,
L. The inhibitory collagen receptor LAIR-1 (CD305). _J. Leukoc. Biol._ 83, 799–803 (2008). Article CAS PubMed Google Scholar * Guo, H. F. et al. Pro-metastatic collagen lysyl hydroxylase
dimer assemblies stabilized by Fe(2+)-binding. _Nat. Commun._ 9, 512 (2018). Article PubMed PubMed Central CAS Google Scholar * Levental, K. R. et al. Matrix crosslinking forces tumor
progression by enhancing integrin signaling. _Cell_ 139, 891–906 (2009). Article CAS PubMed PubMed Central Google Scholar * Gilkes, D. M. et al. Procollagen lysyl hydroxylase 2 is
essential for hypoxia-induced breast cancer metastasis. _Mol. Cancer Res._ 11, 456–466 (2013). Article CAS PubMed PubMed Central Google Scholar * Gilkes, D. M., Bajpai, S., Chaturvedi,
P., Wirtz, D. & Semenza, G. L. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in
fibroblasts. _J. Biol. Chem._ 288, 10819–10829 (2013). Article CAS PubMed PubMed Central Google Scholar * Heikkinen, J. et al. Lysyl hydroxylase 3 is a multifunctional protein
possessing collagen glucosyltransferase activity. _J. Biol. Chem._ 275, 36158–36163 (2000). Article CAS PubMed Google Scholar * Scietti, L. et al. Molecular architecture of the
multifunctional collagen lysyl hydroxylase and glycosyltransferase LH3. _Nat. Commun._ 9, 3163 (2018). Article PubMed PubMed Central CAS Google Scholar * Hamazaki, H. & Hamazaki, M.
H. Catalytic site of human protein-glucosylgalactosylhydroxylysine glucosidase: Three crucial carboxyl residues were determined by cloning and site-directed mutagenesis. _Biochem. Biophys.
Res. Commun._ 469, 357–362 (2016). Article CAS PubMed Google Scholar * Mercer, D. K., Nicol, P. F., Kimbembe, C. & Robins, S. P. Identification, expression, and tissue distribution
of the three rat lysyl hydroxylase isoforms. _Biochem. Biophys. Res. Commun._ 307, 803–809 (2003). Article CAS PubMed Google Scholar * Yeowell, H. N. & Walker, L. C. Tissue
specificity of a new splice form of the human lysyl hydroxylase 2 gene. _Matrix Biol._ 18, 179–187 (1999). Article CAS PubMed Google Scholar * Yeowell, H. N., Walker, L. C., Mauger, D.
M., Seth, P. & Garcia-Blanco, M. A. TIA nuclear proteins regulate the alternate splicing of lysyl hydroxylase 2. _J. Investig. Dermatol._ 129, 1402–1411 (2009). Article CAS PubMed
Google Scholar * Venables, J. P. et al. MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. _Nat. Commun._ 4, 2480 (2013).
Article PubMed CAS Google Scholar * Braeutigam, C. et al. The RNA-binding protein Rbfox2: an essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion.
_Oncogene_ 33, 1082–1092 (2014). Article CAS PubMed Google Scholar * Shapiro, I. M. et al. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular
phenotype. _PLoS Genet_ 7, e1002218 (2011). Article CAS PubMed PubMed Central Google Scholar * Schliekelman, M. J. et al. Targets of the tumor suppressor miR-200 in regulation of the
epithelial-mesenchymal transition in cancer. _Cancer Res_. 71, 7670–7682 (2011). Article CAS PubMed PubMed Central Google Scholar * Yang, Y. et al. The Notch ligand Jagged2 promotes
lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. _J. Clin. Investig._ 121, 1373–1385 (2011). Article CAS PubMed PubMed Central Google Scholar * Chen, L. et
al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. _Nat. Commun._ 5, 5241 (2014). Article CAS PubMed Google
Scholar * Chen, L. et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. _Cancer Disco._ 8, 1156–1175 (2018). Article CAS Google Scholar *
Banushi, B. et al. Regulation of post-Golgi LH3 trafficking is essential for collagen homeostasis. _Nat. Commun._ 7, 12111 (2016). Article CAS PubMed PubMed Central Google Scholar *
Jurgensen, H. J. et al. A novel functional role of collagen glycosylation: interaction with the endocytic collagen receptor uparap/ENDO180. _J. Biol. Chem._ 286, 32736–32748 (2011). Article
PubMed PubMed Central CAS Google Scholar * Sricholpech, M. et al. Lysyl hydroxylase 3-mediated glucosylation in type I collagen: molecular loci and biological significance. _J. Biol.
Chem._ 287, 22998–23009 (2012). Article CAS PubMed PubMed Central Google Scholar * Schneider, V. A. & Granato, M. The myotomal diwanka (lh3) glycosyltransferase and type XVIII
collagen are critical for motor growth cone migration. _Neuron_ 50, 683–695 (2006). Article CAS PubMed Google Scholar * Katzman, R. L., Kang, A. H. & Beachey, E. H. Collagen-induced
platelet aggregation: involement of an active glycopeptide fragment (alpha1-CB5). _Science_ 181, 670–672 (1973). Article CAS PubMed Google Scholar * Tasker, P. N. et al. Association of
PLOD1 polymorphisms with bone mineral density in a population-based study of women from the UK. _Osteoporos. Int_. 17, 1078–1085 (2006). Article CAS PubMed Google Scholar * Mumm, S. et
al. Bruck syndrome 2 variant lacking congenital contractures and involving a novel compound heterozygous PLOD2 mutation. _Bone._ 130, 115047 (2020). Article CAS PubMed Google Scholar *
Goveia, J. et al. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. _Cancer Cell_ 37, 21–36 (2020).
e13. Article CAS PubMed Google Scholar * Chen, Y. et al. Lysyl Hydroxylase 2 Is Secreted by Tumor Cells and Can Modify Collagen in the Extracellular Space. _J. Biol. Chem._ 291,
25799–25808 (2016). Article CAS PubMed PubMed Central Google Scholar * Albritton, J. L. et al. Ultrahigh-throughput Generation and Characterization of Cellular Aggregates in
Laser-ablated Microwells of Poly(dimethylsiloxane). _RSC Adv._ 6, 8980–8991 (2016). Article CAS PubMed Google Scholar * Guo, H. F. et al. A scalable lysyl hydroxylase 2 expression system
and luciferase-based enzymatic activity assay. _Arch. Biochem. Biophys._ 618, 45–51 (2017). Article CAS PubMed PubMed Central Google Scholar * Waterhouse, A. et al. SWISS-MODEL:
homology modelling of protein structures and complexes. _Nucleic Acids Res_. 46, W296–W303 (2018). Article CAS PubMed PubMed Central Google Scholar * Yamauchi, M., Terajima, M. &
Shiiba, M. Lysine Hydroxylation and Cross-Linking of Collagen. _Methods Mol. Biol._ 1934, 309–324 (2019). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This work
was supported in part by National Institutes of Health grants R01CA105155 (J.M.K. and M.Y.) and K99CA225633 (H-F.G.); Cancer Prevention and Research Institute of Texas (CPRIT) grant
RP160652 (J.M.K); and the Gloria Lupton Tennison Distinguished Professorship in Lung Cancer (J.M.K.). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Thoracic/Head and Neck
Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA Hou-Fu Guo, Neus Bota-Rabassedas, B. Leticia Rodriguez, Don L. Gibbons, Yulong Chen, Priyam Banerjee,
Xiaochao Tan, Xin Liu, Jiang Yu & Jonathan M. Kurie * Division of Oral and Craniofacial Health Sciences, Adams School of Dentistry, University of North Carolina at Chapel Hill, Chapel
Hill, NC, USA Masahiko Terajima & Mitsuo Yamauchi * Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA Chi-Lin Tsai,
Xiaoyan Wang & John A. Tainer * Institute for Human Health & Disease Intervention (I-HEALTH) and Department of Chemistry & Biochemistry, Florida Atlantic University, Jupiter, FL,
USA Michal Tokmina-Roszyk, Roma Stawikowska & Gregg B. Fields * Department of Biosciences, Rice University, Houston, TX, USA Mitchell D. Miller & George N. Phillips Jr * Division of
Medicinal Chemistry, Targeted Therapeutic Drug Discovery and Development Program, College of Pharmacy, The University of Texas at Austin, Austin, TX, USA Juhoon Lee & Kevin N. Dalby *
Division of Chemical Biology & Medicinal Chemistry, College of Pharmacy, The University of Texas at Austin, Austin, TX, USA Juhoon Lee & Kevin N. Dalby * Department of Medicine, Dan
L. Duncan Cancer Center, Baylor College of Medicine, Houston, TX, USA Chad J. Creighton * Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer
Center, Houston, TX, USA Chad J. Creighton * Department of Chemistry, Rice University, Houston, TX, USA George N. Phillips Jr Authors * Hou-Fu Guo View author publications You can also
search for this author inPubMed Google Scholar * Neus Bota-Rabassedas View author publications You can also search for this author inPubMed Google Scholar * Masahiko Terajima View author
publications You can also search for this author inPubMed Google Scholar * B. Leticia Rodriguez View author publications You can also search for this author inPubMed Google Scholar * Don L.
Gibbons View author publications You can also search for this author inPubMed Google Scholar * Yulong Chen View author publications You can also search for this author inPubMed Google
Scholar * Priyam Banerjee View author publications You can also search for this author inPubMed Google Scholar * Chi-Lin Tsai View author publications You can also search for this author
inPubMed Google Scholar * Xiaochao Tan View author publications You can also search for this author inPubMed Google Scholar * Xin Liu View author publications You can also search for this
author inPubMed Google Scholar * Jiang Yu View author publications You can also search for this author inPubMed Google Scholar * Michal Tokmina-Roszyk View author publications You can also
search for this author inPubMed Google Scholar * Roma Stawikowska View author publications You can also search for this author inPubMed Google Scholar * Gregg B. Fields View author
publications You can also search for this author inPubMed Google Scholar * Mitchell D. Miller View author publications You can also search for this author inPubMed Google Scholar * Xiaoyan
Wang View author publications You can also search for this author inPubMed Google Scholar * Juhoon Lee View author publications You can also search for this author inPubMed Google Scholar *
Kevin N. Dalby View author publications You can also search for this author inPubMed Google Scholar * Chad J. Creighton View author publications You can also search for this author inPubMed
Google Scholar * George N. Phillips Jr View author publications You can also search for this author inPubMed Google Scholar * John A. Tainer View author publications You can also search for
this author inPubMed Google Scholar * Mitsuo Yamauchi View author publications You can also search for this author inPubMed Google Scholar * Jonathan M. Kurie View author publications You
can also search for this author inPubMed Google Scholar CONTRIBUTIONS J.M.K. and H-F.G. conceived the study. J.M.K. oversaw the work of H-F.G., N.B-R., Y.C., X.L., J.Y., and X.T. H-F.G.
designed, performed, and analyzed amino acid sequence alignment and LH and GLT enzymatic activity assays. M.T. and M.Y. designed, performed, and analyzed the collagen cross-linking assays.
N.B-R and P.B. designed, performed, and analyzed the studies on 344SQ multicellular aggregates co-cultured on MC cells. B.L.R. and D.L.G performed and analyzed the flow cytometry of immune
cell populations. Y.C. and X.T. created expression vectors and performed transfections on MC cells. C-L.T., J.A.T, M.D.M., and G.N.P. assisted with interpretation of protein crystallographic
analyses that were removed during manuscript revision. M.T-R., R.S., H-F.G., and G.B.F. prepared substrates utilized in enzymatic assays. X.L. and J.Y. performed mouse breeding, tumor cell
injections, and necropsies of orthotopic tumor-bearing mice. K.N.D and J.L contributed to the homology model of LH2 and telopeptide that was not included in the paper. CORRESPONDING AUTHOR
Correspondence to Jonathan M. Kurie. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION PEER REVIEW FILE SUPPLEMENTARY INFORMATION DESCRIPTION OF ADDITIONAL
SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which
permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to
the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless
indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or
exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints
and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Guo, HF., Bota-Rabassedas, N., Terajima, M. _et al._ A collagen glucosyltransferase drives lung adenocarcinoma progression in mice.
_Commun Biol_ 4, 482 (2021). https://doi.org/10.1038/s42003-021-01982-w Download citation * Received: 05 June 2020 * Accepted: 08 March 2021 * Published: 19 April 2021 * DOI:
https://doi.org/10.1038/s42003-021-01982-w SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative