Genomic epidemiology of animal-derived tigecycline-resistant escherichia coli across china reveals recent endemic plasmid-encoded tet(x4) gene
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Public health interventions to control the recent emergence of plasmid-mediated tigecycline resistance genes rely on a comprehensive understanding of its epidemiology and
distribution over a wide range of geographical scales. Here we analysed an _Escherichia coli_ collection isolated from pigs and chickens in China in 2018, and ascertained that the _tet_(X4)
gene was not present at high prevalence across China, but was highly endemic in northwestern China. Genomic analysis of _tet_(X4)-positive _E. coli_ demonstrated a recent and regional
dissemination of _tet_(X4) among various clonal backgrounds and plasmids in northwestern China, whereas a parallel epidemic coincided with the independent acquisition of _tet_(X4) in _E.
coli_ from the remaining provinces. The high genetic similarity of _tet_(X4)-positive _E. coli_ and human commensal _E. coli_ suggests the possibility of its spreading into humans. Our study
provides a systematic analysis of the current epidemiology of _tet_(X4) and identifies priorities for optimising timely intervention strategies. SIMILAR CONTENT BEING VIEWED BY OTHERS
EMERGENCE OF TRANSFERABLE TIGECYCLINE AND ERAVACYCLINE RESISTANCE GENE _TET_(X4) IN _ESCHERICHIA COLI_ ISOLATES FROM IRAN Article Open access 13 May 2025 DYNAMICS OF EXTENDED-SPECTRUM
CEPHALOSPORIN RESISTANCE GENES IN _ESCHERICHIA COLI_ FROM EUROPE AND NORTH AMERICA Article Open access 12 December 2022 POPULATION STRUCTURE AND ANTIBIOTIC RESISTANCE OF SWINE
EXTRAINTESTINAL PATHOGENIC _ESCHERICHIA COLI_ FROM CHINA Article Open access 10 July 2024 INTRODUCTION The incessant emergence of novel and transmissible antimicrobial resistance (AMR)
mechanisms has frustrated clinicians because limited antimicrobial agents are left for the treatment of infectious diseases1. Classified by the World Health Organization as a critically
important antimicrobial agent, tigecycline provides a key line of defense against multi-drug-resistant bacteria, particularly in cases of life-threatening infections2. Up until 2019,
reported resistance to tigecycline was commonly mediated by mutational and regulatory changes with limited lateral transferability3,4. However, plasmid-encoded _tet_(X) genes that confer
high-level tigecycline resistance were first described in isolates from animals and humans in China in around 20185,6,7. Subsequently, several more _tet_(X) variants—_tet_(X3), _tet_(X4),
and _tet_(X5)—were identified on transferable plasmids in _Enterobacteriaceae_ and _Acinetobacter_ isolates from animals and humans in China5,6. In addition to their ability to degrade all
tetracyclines, the novel plasmid-mediated _tet_(X) variants show a much more efficient horizontal transferability, making the host bacteria and recipient pathogens a new and severe threat to
global public health5,6,7. Initial epidemiological studies in China indicated that the current presence of _tet_(X3) and _tet_(X4) is still rare in the human sector, but more common in food
animals, meat products, and the surrounding niches5,6,7,8, suggesting that the food animals may be large reservoir of these novel mobile tigecycline resistance genes. A further
retrospective study reported that the emergence of _tet_(X4) in food animals is a recent event9, but is now spreading geographically5,6,7,8,10, indicating an emerging risk of food animals as
reservoirs in spreading these genes. Public health interventions to control the spread of AMR genes rely on a comprehensive understanding of its current epidemiology and distribution over a
wide range of geographical scales. However, tigecycline is not authorized in food animals, its resistance in animals is not routinely monitored and largely unknown. Given the clinical
importance of tigecycline and the emerging risk of food animals in spreading its resistance, a further comprehensive surveillance is urgently recommended to underline the current situation
of these newly identified _tet_(X) variants5,6,7,8,9,10. Here, we analysed an _E. coli_ collection isolated from pigs and chickens in China 2018. We ascertained that the gene _tet_(X4) was
not present in high prevalence on a China-wide scale, but was highly endemic in northwestern China. We illustrated a complex combination of multiple genetic vehicles (mobile genetic
elements, plasmids and bacterial lineages) in the spreading of the _tet_(X4) gene, and showed a possibility of _tet_(X4)-positive _E. coli_ in spreading into the humans. RESULTS _TET_(X4)
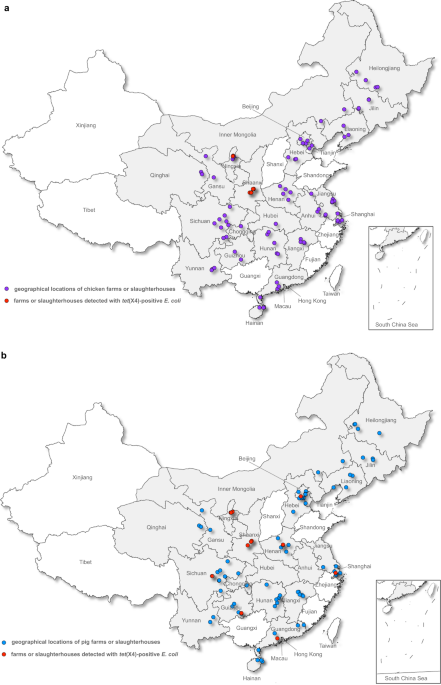
WAS ENDEMIC IN NORTHWESTERN CHINA From the 2475 _E. coli_ isolates included in the study, 125 tigecycline-non-susceptible _E. coli_ were recovered. The _tet_(X4) gene was detected in 95
isolates originating from eight provinces, including 60 pig isolates (60/1,230, 4.9%, 95% confidence interval (CI): 3.7–6.2%) and 35 chicken isolates (35/1,245, 2.8%, 95% CI: 2.0–3.9%)
(Table 1 and Fig. 1). Shaanxi and Ningxia, two neighbouring provinces in northwestern China, had a relatively high _tet_(X4) prevalence in both pigs (30/60, 50.0%, Shaanxi; 20/45, 44.4%,
Ningxia) and chickens (20/45, 44.4%, Shaanxi; 15/45, 33.3%, Ningxia). In contrast, the _tet_(X4) gene appeared sporadically among pig isolates from Sichuan (4/150, 2.7%), Henan (2/75, 2.7%),
Guizhou (1/60, 1.7%), Beijing (1/135, 0.74%), Shanghai (1/75, 1.3%), and Guangdong (1/75, 1.3%) and was negative in the remaining 14 provinces. The chicken isolates that were positive for
_tet_(X4) were only detected from Shaanxi and Ningxia (Table 1 and Fig. 1). All amplified fragments had 100% sequence homology to the original _tet_(X4) gene5. The _tet_(X3) and _tet_(X5)
genes were absent in the current strain collection. The underlying tigecycline resistance mechanisms of the 30 tigecycline-non-susceptible _E. coli_ that were all negative for novel _tet_(X)
variants were not explored in the current study and will be discussed elsewhere. THE ANTIMICROBIAL RESISTANT PHENOTYPE AND GENOTYPE Susceptibility testing confirmed that 95
_tet_(X4)-positive _E. coli_ showed resistance to tigecycline (minimum inhibitory concentrations (MICs) 4–32 mg L−1), doxycycline (32–128 mg L−1), and florfenicol (32–128 mg L−1) but
exhibited sensitivity to meropenem. A few of the isolates showed resistance to colistin (_n_ = 3, 3.2%), cefepime (_n_ = 1, 1.1%), ceftriaxone (_n_ = 4, 4.2%), aztreonam (_n_ = 5, 5.3%), and
gentamicin (_n_ = 7, 7.4%), while the majority were resistant to ampicillin (_n_ = 92, 96.8%) and amoxicillin-clavulanate (_n_ = 94, 98.8%) (Supplementary Data 1). Apart from _tet_(X4), a
median of 13 AMR genes (range, 2–20) was detected in the genome of each isolate. Genes coding for resistance to phenicols (_floR_, 91/95), tetracyclines (_tet_(A), 87/95), sulfonamides
(_sul2_, 26/39; _sul3_, 71/95), aminoglycosides (_strA_, 81/95; _strB_, 81/95; _aadA2_, 70/95), and trimethoprims (_drfA12_, 69/95) were highly present in the _E. coli_ harbouring _tet_(X4).
The colistin resistance gene _mcr-1_ was detected in two of the 95 _E. coli_ isolates (Supplementary Fig. 1 and Supplementary Data 2). All _tet_(X4)-positive _E. coli_ were classified into
five phylogroups (A, _n_ = 79; B1, _n_ = 12; B2, _n_ = 1; C, _n_ = 1, and F, _n_ = 2). One pig strain SC-P337 (Sichuan, ST4541, O146:H28) was placed into group B2, which is commonly known as
being highly pathogenic, and carried a greater number of virulence-factor-associated genes (_n_ = 62) than isolates from group A (Supplementary Fig. 1 and Supplementary Data 2). GENOMIC
DIVERSITY OF _TET_(X4)-POSITIVE _E. COLI_ In silico multilocus sequence typing (MLST) clustered the 95 _tet_(X4)-positive _E. coli_ into 19 distinct sequence types (STs), with 60 pig
isolates clustered in 18 STs and 35 chicken isolates in seven STs (Supplementary Fig. 2). Although diversified in STs, the majority (76.8%, 73/95) of the isolates were concentrated into five
major STs, 6704 (27/95), 2035 (13/95), 48 (11/95), 1602 (11/95), and 877(11/95), which mostly (73/74) originated from Shaanxi and Ningxia (Fig. 2 and Supplementary Fig. 2). Isolates within
each major ST were characterized by low levels of core genome diversity that was reflected by a relatively limited number of single nucleotide polymorphisms (SNPs) (differing in 3–931 SNPs)
(Fig. 2). However, isolates (_n_ = 10) from the remaining six provinces and municipalities showed relatively greater genetic diversity among each other, composing 8 of the 19 STs (Fig. 2).
VERSATILE PLASMIDS HARBOURING _TET_(X4) Based on the phylogenetic analysis, around one-third of the isolates within each of the major lineages were selected (_n_ = 25), together with most of
the remaining isolates (_n_ = 17), for Oxford Nanopore Technologies (ONT) long-read sequencing (Supplementary Data 2). Hybrid de novo assembly produced 39 completely sequenced plasmids
possessing _tet_(X4) and three circular intermediates (CIs) harbouring _tet_(X4) but without replicons. Incompatibility typing of the 39 plasmids (ranging in sizes from 8581 to 322,239 bp)
showed a striking variety of plasmid types. In addition to a handful of IncX1 plasmids (_n_ = 13; 21,050–82,762 bp) and two untypable plasmids (8581 and 12,805 bp), a majority of the
plasmids (25/39) were detected with multi-replicons, including IncX1-IncN (_n_ = 18; 55,530–71,790 bp), IncX1-IncR (_n_ = 2; 76,546 bp and 77,171 bp), IncX1-IncFIA/B-IncY (_n_ = 2; 133,595
bp and 133,595 bp), IncX1-IncFIA/B-IncHI1A/B (_n_ = 1; 260,533 bp), and IncFIA/B-IncHI1A/B (_n_ = 1; 323,762 bp) (Fig. 2 and Supplementary Data 2). Plasmids of the same Inc type could be
identified not only from isolates of the same lineage, but also from isolates of the different lineages, indicating the spread of the _tet_(X4) gene is combining vertical and horizontal
transferability (Fig. 2). Outside northwestern China, plasmids exhibited limited comparability but endemism to those harboured in the major lineages, such as the two large multi-replicon
plasmids in the isolates from Beijing (ST744, IncFIA/B-IncHI1A/B) and Guangdong (ST10, IncX-IncFIA/B-IncHI1A/B), as well as the small untypable plasmids from Sichuan (ST792) and Guizhou
(ST48); however, the endemic IncX1 and IncX1-IncN plasmids from northwestern China were not observed elsewhere (Fig. 2). MGE ARRANGEMENTS IN SPREADING _TET_(X4) To further analyse the
transfer of _tet_(X4) via mobile genetic elements (MGE), the sequenced plasmids were probed for _tet_(X4) genetic contexts and were subsequently correlated to plasmid types and the host
strains. The genetic environments of _tet_(X4) were clustered into 26 types, differing mostly in the variable context downstream of _tet_(X4) but showing a relatively conserved architecture
upstream of the gene (Fig. 3). Compared with the flanking stance of the two IS_CR2_ copies in the original _tet_(X4)-harbouring plasmid p47EC (IS_CR2_-ORF2-_abh_-_tet_(X4)-IS_CR2_)5, the
upstream copy of IS_CR2_ adjacent to _abh_-_tet_(X4) was absent in most of the present types (20/26) but generally replaced by a truncated IS_1B_ element (IS_1B_-_abh_-_tet_(X4)-IS_CR2_). In
contrast, the downstream copy of IS_CR2_ remained tightly associated with _tet_(X4), forming a unified central region _abh_-_tet_(X4)-IS_CR2_ (Fig. 3). A further downstream search generally
found a second copy of IS_CR2_ (12/26, type 1–3, 6–12, 14, and 19), which was joined by the type IV secretion system component _virD2_ relaxase and led to a set of other resistance
determinants, mainly a gene cluster of _floR_, _tet_(A), _strB_, _strA_, _sul2_, _bla_TEM-1B, and occasionally _mef_(B) and _ble_. Whenever the second copy of IS_CR2_ was absent_, virD2_ and
the adherent resistance genes were linked directly to the first copy of IS_CR2_ downstream of _tet_(X4) (type 4, 5, 13, 24, and 26) (Fig. 3). By connecting the derived genetic environment
types back to plasmids (seven Inc types) and the host strains (16 STs), we illustrated a complex combination of multiple genetic vehicles in the spreading of the _tet_(X4) gene (Fig. 4). THE
PLASMID RESISTOME IN CONNECTING _TET_(X4) Given the fact of co-selection in driving plasmids’ persistence, we further examined the resistome of _tet_(X4)-harbouring plasmids. A median of
eight different AMR genes (range, 1–13) was detected within the 39 completely sequenced plasmids, while the median number of AMR genes was 14 (range, 3–20) when scrutinizing the 39 whole
genomes. Indeed, we found that a majority of _tet_(X4)-positive plasmids (23/39) possessed over half of the whole genomes’ resistance genes (Fig. 5), indicating the central role of
_tet_(X4)-positive plasmids in facilitating the host bacteria’s resistance to antimicrobials. The phenicol resistance gene _floR_ represented the highest occurrence (31/39), followed by
genes conferring resistance to tetracyclines (_tet_(A), 28/39), aminoglycosides (_aph(3”)-Ib_, 27/41 and _aph(6)-Id_, 27/39), sulfonamides (_sul2_, 26/39), and β-lactams (_bla_TEM-1A, 25/39
and _bla_TEM-1B, 18/39). Generally, high numbers of resistance genes were observed in the syncretic plasmids with three or more replicons (median, 11) (Fig. 5 and Supplementary Data 2). HIGH
GENETIC PROPENSITY OF _TET_(X4) IN SPREADING INTO HUMANS To determine the genetic propensity of _tet_(X4)-positive isolates to spread into the human sector, we analysed the genetic
relatedness of our 95 _tet_(X4)-positive _E. coli_ to 287 publicly available draft genomes of human commensal _E. coli_ (PRJNA400107) that were negative for _tet_(X4). The negative isolates
were non-duplicates and originated from healthy individuals across China in 2016 as part of a previous study characterising _mcr-1_ carriage in humans11. The 95 _tet_(X4)-harbouring _E.
coli_ were phylogenetically clustered into three of the four Bayesian lineages generated from the human isolates (L1, _n_ = 3; L3, _n_ = 26, and L4, _n_ = 66) (Fig. 6a). A large proportion
of our animal-derived _tet_(X4)-harbouring _E. coli_ displayed high genetic similarity (25/95, as small as 39 SNPs) to the _E. coli_ of human origin (Fig. 6b) from various provinces
(Supplementary Fig. 3). Of particular concern is that the pig isolate from Shaanxi differed by only 119 SNPs from the human _E. coli_ isolated in this province. DISCUSSION Data remain scarce
on the newly identified plasmid-mediated tigecycline resistance genes. Combining a national _E. coli_ collection and advances in bioinformatics, the present study reveals a comprehensive
understanding of the _tet_(X4) gene in _E. coli_ from food-producing animals across China. It is evident from our data that the _tet_(X4) gene was present at low prevalence at the national
scale in 2018, but was highly endemic in northwestern China. The observed limited genetic diversity of the isolates from northwestern China and the promiscuous plasmids carrying _tet_(X4)
indicate recent and regional dissemination and variably successful clonal backgrounds, thus further extending our previous commentary describing the recent emergence of the _tet_(X4) gene in
China9. Although scattered _tet_(X4)-positive isolates were identified in the remaining six provinces, the distinct STs and plasmids observed suggest that a parallel epidemic has coincided
with the independent acquisition of _tet_(X4)-harbouring plasmids. Given the extensive food animal husbandry and free trade within China, these _tet_(X4)-harbouring “sparks” are presumably
spread over a wide range of geographical scales, as exemplified by the clonal and lateral expansions in northwest China. In this context, it is worrisome to speculate that a national
pandemic of tigecycline-resistant _E. coli_ in food animals is only a matter of time unless there is timely and effective eradication of these “sparks” before the “prairie fire”. Moreover,
our concerns are escalated by the high genetic propensity of _tet_(X4)-positive _E. coli_ for spreading into humans. Although the phylogenetic analysis was based on human _E. coli_ that
positive for _mcr-1_, there were reports of _E. coli_ coharbouring _tet_(X4) and _mcr-1_6,9,12. The high genetic similarity of _E. coli_ between pigs and humans in the endemic region
(Shaanxi province) indicates a possibility of spillover of the _tet_(X4) gene from animals to humans. In this circumstance, in vivo transfer of the _tet_(X4) gene into human pathogens would
be highly likely to occur. This process might be co-selected and magnified by extensively used antibiotics in humans, such as β-lactams and aminoglycosides, as their resistant genes were
observed in this study frequently accompanied by _tet_(X4). The _tet_(X4)-harbouring plasmids resided in a wide variety of ST clones (Fig. 2 and Supplementary Fig. 2), but only three _E.
coli_ Bayesian lineages were implicated (Fig. 6a), thus suggesting a considerably narrow host spectrum of the plasmids carrying this gene. This is likely a similar scenario reminiscent of
_mcr-1_, which resided primarily on IncX4 plasmids that flowed among four lineages (although diversified in 135 STs) of _E. coli_ across China11, but it contrasts with the transmission of
the _bla_NDM gene among eight distinct _E. coli_ lineages in a single Chinese poultry production chain13. However, the _tet_(X4) gene was identified in a large proportion of multireplicon
plasmids, which are diversified in replicon gene combination. This is unusual, but not without precedent in that _mcr-1_ has already been found in several hybrid versions of
plasmids11,14,15, suggesting the involvement of plasmid fusion in the spreading of _tet_(X4). The presence of multiple replicons in _tet_(X4)-positive plasmids might prevent plasmid
incompatibility16 and facilitate their interaction with a broad range of hosts17. Our plasmid resistome analysis confirmed that _tet_(X4)-positive plasmids are indeed versatile in spreading
AMR genes. Further dissemination of these “super plasmids” into clinical settings would be a big threat. However, it remains to be seen whether the plasmids carrying _tet_(X4) are more prone
to hybridisation, but the host ranges, the capacity to capture AMR genes, and the evolution of these new _tet_(X4) genetic platforms will need to be closely monitored. Apart from a certain
documented genetic environment of _tet_(X4), we have here expanded its habitation to a broader territory. IS_CR2_ was found either flanked at both ends of _abh_-_tet_(X4) or existing solely
downstream of _tet_(X4). Different from the transposon paradigm, a single copy of the IS_CR2_ would transpose the adjacent sequences via rolling-circle transposition, which has been
documented in the mobilization of several resistance genes18. In these circumstances, IS_CR2_-mediated transposition might play an essential role in the integration of _tet_(X4) into
multiple plasmids, consequently enlarging the possibility of _tet_(X4) to propagate into the broad host range plasmids as well as other bacterial species. Taken together, the contribution of
MGEs, in particular IS_CR2_, to the spread of _tet_(X4) gene should not be minimised. Despite the findings, this work has several limitations that should be noted. First, we included
approximately 15 _E. coli_ randomly selected from each farm or slaughterhouse, which might not be proportional to the annual production (pig and chickens) of each province or municipality.
Second, the direct screening of _tet_(X4) from _E. coli_ identified without targeted tigecycline phenotype and enrichment steps might underestimate the true prevalence and diversity of _E.
coli_ carrying _tet_(X4), as previously seen in the detection of _mcr-1_11. Third, the majority of the _tet_(X4)-positive _E. coli_ originated from Shaanxi and Ningxia, which might have
biased the deduced results. On a national scale, the current study shows the countenance of the newly emerged _tet_(X4)-mediated tigecycline resistance, which is undeniably a great public
health threat. Based on these results, the intervention priorities should focus on (1) the rapid and thorough eradication of this gene in food animals in northwest China, (2) tight
monitoring of this gene in humans, particularly in people who are epidemiologically related to northwestern China, and (3) continuous national surveillance and risk assessment of this gene
in a broader One Health approach. Furthermore, our results may also serve as a benchmark for the current status of _tet_(X4)-positive _E. coli_ at an early stage, to which future survey data
can be compared, thus facilitating not only the understanding of the paradigmatic shift of resistance, but also critical intervention improvements. METHODS STRAIN COLLECTIONS This study is
based on _E. coli_ collections from China’s national AMR surveillance programme in zoonotic bacteria. The _E. coli_ isolates were collected and submitted by provincial laboratories or
institutes certified as part of the surveillance programme. The participation of farms and slaughterhouses in the surveillance system in each province was on a voluntary basis. Commensal _E.
coli_ isolates were identified without any targeted AMR phenotypes from healthy chickens (cloacal swabs) and pigs (faecal swabs). As of July 2019, the system had received a total of 3124
_E. coli_ isolates in 2018 originating from 233 farms and slaughterhouses across 22 of the 34 provinces and municipalities in China. Because there was a large variation in the number of
isolates from each farm and slaughterhouse, we randomly selected about 15 _E. coli_ isolates from each farm or slaughterhouse in order to minimise the sample bias in the surveillance data,
and this resulted in a total of 2,475 _E. coli_ isolated from 166 farms and slaughterhouses distributed across the country (Fig. 1 and Table 1). Since no live vertebrates were used in this
study, the included _E. coli_ collection is exempt from the IACUC approval process. PCR-BASED SCREENING OF NOVEL _TET_(X) VARIANTS _E. coli_ isolates were inoculated on MacConkey plates
supplemented with tigecycline (2 mg/mL), and the resulting tigecycline-non-susceptible isolates were then subjected to _tet_(X) and variant screening using a two-step PCR analysis. A
universal primer pair (_tet_(X) forward 5′-CCG TTG GAC TGA CTA TGG C-3′; _tet_(X) reverse 5′-TCA ACT TGC GTG TCG GTA A-3′) targeting a 475 bp conserved region of _tet_(X) and the four
_tet_(X) variants was used, and the positives were further screened for _tet_(X3), _tet_(X4), and _tet_(X5) using primer pairs as previously described5,7. The genomic DNA of
_tet_(X3)-carrying _Acinetobacter baumannii_ 34AB, _tet_(X4)-carrying _E. coli_ 47EC, and _tet_(X5)-carrying _A. baumannii_ AB17H194 from previous studies were used as positive controls5,7.
ANTIMICROBIAL SUSCEPTIBILITY TESTING MICs were determined for all _tet_(X4)-carrying _E. coli_ using broth microdilution in Mueller-Hinton broth (Oxoid, Basingstoke, UK). The tested
antibiotics included tigecycline, colistin, meropenem, cefepime, ceftriaxone, ampicillin, amoxicillin-clavulanate, aztreonam, ciprofloxacin, gentamicin, doxycycline, and florfenicol.
Susceptibility was determined according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST, version 10.0, for tigecycline, colistin, and florfenicol)19 and the
Clinical and Laboratory Standards Institute document (CLSI, M100-S29, for the remaining antibiotics)20. Reference strain _E. coli_ ATCC 25922 and _Staphylococcus aureus_ ATCC 29213 served as
the quality control strains. WHOLE-GENOME SEQUENCING AND ASSEMBLY All positive _E. coli_ isolates were subjected to whole-genome sequencing. Briefly, a KAPA Hyper Prep Kit (Kapa Biosystems,
Boston, MA, US) was used for library construction, and 300-bp paired-end reads with a minimum of 150-fold coverage for each isolate were obtained following sequencing using the Illumina
HiSeq X Ten System. A draft assembly of the cleaned reads was generated using SPAdes version 3.9.021. To determine the plasmid or chromosome location and the genetic environment of the
_tet_(X4) gene, a number of isolates were selected based on phylogenetic analysis and background information regarding the source for further sequencing by ONT long-read sequencing. Briefly,
libraries were prepared using the Rapid Barcoding Kit (SQK-RBK004) and subjected to ONT long-read sequencing in R9.4.1 flow cells in a MinION sequencer according to the standard protocol A
0.3.4. The resulting long reads were subjected to hybrid de novo assembly in combination with Illumina short reads22. PHYLOGENETIC ANALYSIS Core-genome SNP-based Neighbour-Joining
phylogenetic trees were constructed for all sequenced isolates using Parsnp in the Harvest package23 with default parameter settings, and these were visualised using iTOL v524 with the
corresponding features of each isolate. To determine the genetic plasticity of the _tet_(X4) gene in its spread into the humans, we analysed the genetic relatedness of all _tet_(X4)-positive
_E. coli_ in the current study together with 287 publicly available draft genomes of _E. coli_ (PRJNA400107) that are negative for _tet_(X4). The negative isolates were non-duplicates and
originated from human stool samples collected from healthy individuals in 2016 across China, in a previous study characterising _mcr-1_ carriage in humans11. A core-genome SNP tree was
constructed and visualised, as described above. IDENTIFICATION OF STS, AMR, AND VIRULENCE GENES Multi-locus sequence types (MLST) were assigned according to the _E. coli_ MLST database25 by
mapping cleaned reads to the alleles using SRST226. A minimum spanning tree based on generated sequence types was constructed in BioNumerics version 7.0 (Applied Maths) and used to
differentiate the isolates in terms of their origin. Given the clinical importance of AMR and virulence in _E. coli_, a targeted analysis of all known acquired AMR genes and
virulence-factor-associated genes was performed within our genomic dataset. These genes were screened using abricate against the ResFinder 2.127 and vfdb28 databases (>90% identity). The
classification of each strain into phylogenetic groups was performed according to the scheme described previously29. _TET_(X4) GENE LOCATION, ENVIRONMENT AND PLASMID TYPING We determined the
plasmid or chromosome location of the _tet_(X4) gene in the selected isolates by ONT long-read sequencing analysis. Contigs harbouring _tet_(X4) were searched and extracted from the
assemblies using contig-puller (https://github.com/kwongj/contig-puller) and checked for cyclization. By searching against PlasmidFinder30 (>95% identity and >90% coverage), circular
contigs harbouring plasmid replicons were considered plasmid-borne and circular contigs with no replicons detected were considered circular intermediates. All contigs were submitted to
PATRIC31 for the putative coding sequences of the genes flanking _tet_(X4). STATISTICS AND REPRODUCIBILITY Descriptive analysis on prevalence, 95% confidence interval and median calculation
were performed using functions provided in Excel 2019 (Microsoft). Exact sample sizes for each group were described in Table 1. Source data used to plot Figs. 2–5, Supplementary Figs. 1 and
2 are archived in Supplementary Data 2. REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this article. DATA
AVAILABILITY Data supporting the findings of this study are included in this article and in the Supplementary Information. Genome assemblies of the 95 _tet_(X4)-positive _E. coli_ have been
deposited in the NCBI and are registered under BioProject accession no. PRJNA625924. All data are available from the corresponding authors upon reasonable request. REFERENCES * The Review on
Antimicrobial Resistance. _Tackling Drug-Resistant Infections Globally: Final Report and Recommendations_. https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf
(2016). * WHO. _Critically Important Antimicrobials for Human Medicine_. https://apps.who.int/foodsafety/publications/antimicrobials-fifth/en/ (2016). * Sun, Y. et al. The emergence of
clinical resistance to tigecycline. _Int. J. Antimicrob. Ag._ 41, 110–116 (2012). Article Google Scholar * Tuckman, M., Petersen, P. & Projan, S. Mutations in the interdomain loop
region of the _tet_(A) tetracycline resistance gene increase efflux of minocycline and glycylcyclines. _Microb. Drug Resist._ 6, 277–282 (2000). Article CAS Google Scholar * He, T. et al.
Emergence of plasmid mediated high-level tigecycline resistance genes in animals and humans. _Nat. Microbiol._ 4, 1450–1456 (2019). Article CAS Google Scholar * Sun, J. et al.
Plasmid-encoded _tet_(X) genes that confer high-level tigecycline resistance in _Escherichia coli_. _Nat. Microbiol._ 4, 1457–1464 (2019). Article CAS Google Scholar * Wang, L. et al.
Novel plasmid-mediated _tet_(X5) gene conferring resistance to tigecycline, eravacycline and omadacycline in clinical _Acinetobacter baumannii_. _Antimicrob. Agents Ch._ 64, e01326–19
(2019). Google Scholar * Bai, L. et al. Detection of plasmid-mediated tigecycline-resistant gene _tet_(X4) in _Escherichia coli_ from pork, Sichuan and Shandong Provinces, China.
_Eurosurveillance_. 24, 1900340 (2019). * Sun, C. et al. Plasmid-mediated tigecycline-resistant gene _tet_(X4) in _Escherichia coli_ from food-producing animals, China, 2008–2018. _Emerg.
Microbes Infec._ 8, 1524–1527 (2019). Article Google Scholar * Chen, C. et al. Emergence of mobile tigecycline resistance mechanism in _Escherichia coli_ strains from migratory birds in
China. _Emerg. Microbes Infec._ 8, 1219–1222 (2019). Article Google Scholar * Shen, Y. et al. Anthropogenic and environmental factors associated with high incidence of _mcr-1_ carriage in
humans across China. _Nat. Microbiol._ 3, 1054–1062 (2018). Article CAS Google Scholar * He, T. et al. Co-existence of _tet_(X4) and _mcr-1_ in two porcine _Escherichia coli_ isolates.
_J. Antimicrob. Chemother._ 75, 764–766 (2020). Article CAS Google Scholar * Wang, Y. et al. Comprehensive resistome analysis reveals the prevalence of NDM and MCR-1 in Chinese poultry
production. _Nat. Microbiol._ 2, 16260 (2017). Article CAS Google Scholar * Sun, J. et al. Towards understanding MCR-like colistin resistance. _Trends Microbiol._ 26, 794–708 (2018).
Article CAS Google Scholar * Sun, J. et al. Co-transfer of _bla_NDM-5 and _mcr-1_ by an IncX3-X4 hybrid plasmid in _Escherichia coli_. _Nat. Microbiol._ 1, 16176 (2016). Article CAS
Google Scholar * Chen, Z. et al. Characterization of pMC11, a plasmid with dual origins of replication isolated from _Lactobacillus casei_ MCJ and construction of shuttle vectors with each
replicon. _Appl. Microbiol. Biotechnol._ 98, 5977–5989 (2014). Article CAS Google Scholar * Villa, L., Garcıa-Fernandez, A., Fortini, D. & Carattoli, A. Replicon sequence typing of
IncF plasmids carrying virulence and resistance determinants. _J. Antimicrob. Chemother._ 65, 2518–2529 (2010). Article CAS Google Scholar * Toleman, M. A., Bennett, P. M. & Walsh, T.
R. ISCR elements: novel gene-capturing systems of the 21st century? _Microbiol Mol. Biol. Rev._ 70, 296–316 (2006). Article CAS Google Scholar * _European Committee on Antimicrobial
Susceptibility Testing_. Clinical breakpoints for bacteria. EUCAST. http://www.eucast.org/clinical_breakpoints (2019). * CLSI. _Performance standards for antimicrobial susceptibility
testing_. Twentieth Informational Supplement. M100-S29 (Clinical and Laboratory Standards Institute, Wayne, PA, USA, 2019). * Nurk, S. et al. Assembling genomes and mini-metagenomes from
highly chimeric reads. research in computational molecular biology. _Lect. Notes Comput. Sc._ 7821, 158–170 (2013). Article Google Scholar * Wick, R. R., Judd, L. M., Gorrie, C. L. &
Holt, K. E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. _PLoS Comput. Biol._ 13, e1005595 (2017). Article Google Scholar * Treangen, T. J.,
Ondov, B. D., Koren, S. & Phillippy, A. M. The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. _GenomeBiol_ 15, 524–539
(2014). Google Scholar * Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. _Nucleic Acids Res._
44, W242–5 (2016). Article CAS Google Scholar * Aanensen, D. M. & Spratt, B. G. The multilocus sequence typing network: mlst.net. _Nucleic Acids Res._ 33, W728–33 (2005). Article CAS
Google Scholar * Inouye, M. et al. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. _Genome Med._ 6, 90 (2014). Article Google Scholar * Zankari, E.
et al. Identification of acquired antimicrobia resistance genes. _J. Antimicrob. Chemother._ 67, 2640–4 (2012). Article CAS Google Scholar * Liu, B., Zheng, D., Jin, Q., Chen, L. &
Yang, J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. _Nucleic Acids Res._ 47, D687–D692 (2012). Article Google Scholar * Clermont, O., Bonacorsi, S.
& Bingen, E. Rapid and simple determination of the _Escherichia coli_ phylogenetic group. _Appl Environ. Microbiol._ 66, 4555–4558 (2000). Article CAS Google Scholar * Carattoli, A.
et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. _Antimicrob. Agents Chemother._ 58, 3895–3903 (2014). Article Google Scholar *
Wattam, A. R. et al. Improvements to PATRIC, the all bacterial bioinformatics database and analysis resource center. _Nucleic Acids Res._ 45, D535–42 (2017). Article CAS Google Scholar
Download references ACKNOWLEDGEMENTS We thank the researchers involved in the China national surveillance programme on AMR in zoonotic bacteria, and Anette Hulth at Public Health Agency of
Sweden for kindly editing the manuscript. This work was supported in part by the National Key Research and Development Program of China under Grant [2018YFD0500300] and the National Natural
Science Foundation of China under Grants [31930110 and 81991535]. AUTHOR INFORMATION Author notes * These authors contributed equally: Chengtao Sun, Mingquan Cui. AUTHORS AND AFFILIATIONS *
Beijing Key Laboratory of Detection Technology for Animal Food Safety, College of Veterinary Medicine, China Agricultural University, Beijing, China Chengtao Sun, Shan Zhang, Dejun Liu, Bo
Fu, Rina Bai, Yaxin Wang, Jianzhong Shen, Congming Wu & Yang Wang * China Institute of Veterinary Drug Control, Beijing, China Mingquan Cui, Zekun Li, Hejia Wang, Li Song, Chunping
Zhang, Qi Zhao & Shixin Xu Authors * Chengtao Sun View author publications You can also search for this author inPubMed Google Scholar * Mingquan Cui View author publications You can
also search for this author inPubMed Google Scholar * Shan Zhang View author publications You can also search for this author inPubMed Google Scholar * Dejun Liu View author publications You
can also search for this author inPubMed Google Scholar * Bo Fu View author publications You can also search for this author inPubMed Google Scholar * Zekun Li View author publications You
can also search for this author inPubMed Google Scholar * Rina Bai View author publications You can also search for this author inPubMed Google Scholar * Yaxin Wang View author publications
You can also search for this author inPubMed Google Scholar * Hejia Wang View author publications You can also search for this author inPubMed Google Scholar * Li Song View author
publications You can also search for this author inPubMed Google Scholar * Chunping Zhang View author publications You can also search for this author inPubMed Google Scholar * Qi Zhao View
author publications You can also search for this author inPubMed Google Scholar * Jianzhong Shen View author publications You can also search for this author inPubMed Google Scholar * Shixin
Xu View author publications You can also search for this author inPubMed Google Scholar * Congming Wu View author publications You can also search for this author inPubMed Google Scholar *
Yang Wang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS C.T.S., C.M.W., S.X.X., J.Z.S., and Y.W. designed the study and supervised the
whole project. C.T.S., M.Q.C., S.Z., B.F., Z.K.L., R.N.B. and Y.X.W. conducted the screening of tigecycline-non-susceptible _E. coli_. S.Z., B.F., R.N.B. and Y.X.W. conducted the PCR
screening, antimicrobial susceptibility testing, and WGS library construction. C.T.S. interpreted the data and performed all bioinformatics analyses. C.T.S. and M.Q.C. drafted the paper.
L.S., H.J.W., C.P.Z., Q.Z. and D.J.L. gave important inputs to the paper. All authors reviewed, revised, and approved the final paper. CORRESPONDING AUTHORS Correspondence to Shixin Xu or
Congming Wu. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 DESCRIPTION OF
ADDITIONAL SUPPLEMENTARY FILES REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sun, C., Cui, M., Zhang, S. _et al._ Genomic epidemiology of animal-derived tigecycline-resistant _Escherichia coli_ across China reveals
recent endemic plasmid-encoded _tet_(X4) gene. _Commun Biol_ 3, 412 (2020). https://doi.org/10.1038/s42003-020-01148-0 Download citation * Received: 09 February 2020 * Accepted: 14 July 2020
* Published: 31 July 2020 * DOI: https://doi.org/10.1038/s42003-020-01148-0 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative