Population genomic analysis identifies the complex structural variation at the fibromelanosis (fm) locus in chicken
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Phenotypic diversity and its genetic basis are central questions in biology, with domesticated animals offering valuable insights due to their rapid evolution the last 10,000 years.
In chickens, fibromelanosis (FM) is a striking pigmentation phenotype characterized by hyperpigmentation. A previous study identified a complex structural variant involving both two large
duplications (127.4 and 170.5 kb in size) and inversions associated with upregulated expression of the _Endothelin 3_ (_EDN3_) gene. However, the detailed organization of the structural
arrangements have remained unclear. In this study, we conducted a comprehensive genomic survey of 517 FM chickens representing 44 different populations. Our results elucidate the complex
arrangement of the duplications and inversions at the _FM_ locus based on the large-scale genomic survey, population level genotyping, and linkage disequilibrium analysis, providing
conclusive support for one specific configuration of the two large duplications, resolving a controversy that has been unresolved for more than a decade. Our results show that the birth of
this complex structural variant must have involved an interchromosomal rearrangement creating fixed heterozygosity due to sequence differences between the two copies of the 127.4 kb
duplication. This study shows how population genomics can be used to understand complex structural variations that underlie phenotypic variation. SIMILAR CONTENT BEING VIEWED BY OTHERS THREE
CHROMOSOME-LEVEL DUCK GENOME ASSEMBLIES PROVIDE INSIGHTS INTO GENOMIC VARIATION DURING DOMESTICATION Article Open access 11 October 2021 A CHROMOSOME-LEVEL GENOME ASSEMBLY FOR THE SILKIE
CHICKEN RESOLVES COMPLETE SEQUENCES FOR KEY CHICKEN METABOLIC, REPRODUCTIVE, AND IMMUNITY GENES Article Open access 06 December 2023 EVOLUTIONARY ORIGIN OF GENOMIC STRUCTURAL VARIATIONS IN
DOMESTIC YAKS Article Open access 19 September 2023 INTRODUCTION How phenotypic diversity evolves and its genetic basis is a fundamental question in biology. Domesticated animals constitute
a valuable resource to explore this topic due to their rapid phenotypic evolution within the last 10,000 years1. A characteristic feature of domestic animals is altered pigmentation pattern,
a phenotypic change that occurred early during domestications of animals as documented by ancient illustrations and documents. A striking pigmentation phenotype in domestic chicken is
fibromelanosis (FM) that is widespread among Asian chicken breeds, like Chinese Silkie and Ayam Cemani from Indonesia. FM is characterized by a massive expansion of melanocytes resulting in
excessive skin and tissue pigmentation2. The FM trait is highly valued culturally and commercially in Asia. For instance, Silkie chickens are prized in ornamental breeding for their
distinctive appearance, while Ayam Cemani chickens hold cultural significance and command high market prices3. FM chickens are valued for their deep eumelanin deposition, used in food such
as Chinese black-bone soup and in Chinese traditional medicine4,5. A previous study demonstrated that the dominant FM trait in several breeds of chicken is caused by a complex structural
rearrangement involving two duplications, 127.4 and 170.5 kb in size2. One of the duplications encompass the _EDN3_ gene, which has a critical role for melanoblast differentiation and
expansion6. The fact that _EDN3_ shows a highly significant upregulation at the mRNA level in skin from FM chickens strongly suggested that this is the causal mechanism for the massive
expansion of pigment cells in FM chicken2. It was not possible to resolve the organization of the complex rearrangement with the short-read whole genome sequence data available a decade ago,
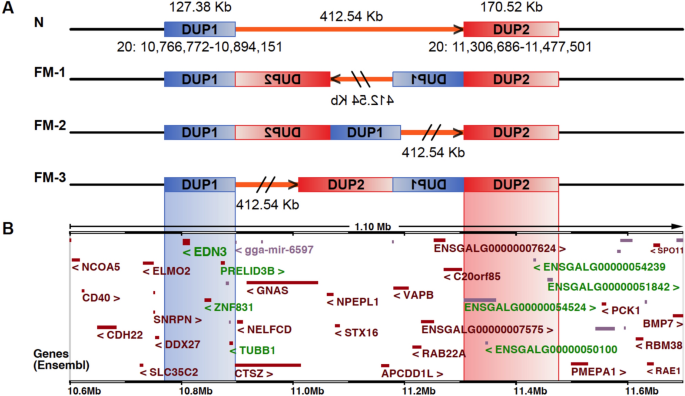
even with the structural variation detection tools widely used today7, and three possible configurations of the complex structural rearrangement (Fig. 1) were established based on PCR
analysis of breakpoint regions2. A single recombinant found in a pedigree segregating for the _FM_ mutation was only consistent with one of the possible configurations denoted FM-22.
However, more recent data based on long read sequencing assemblies have given conflicting results, in which some studies2,8,9 have supported the FM-2 configuration whereas other studies came
to the conclusion that their assembly of this region from Yeonsan Ogye10 and Silkie11 chicken supported the FM-1 configuration. In order to resolve the structural arrangement of the _FM_
locus, here we have analyzed whole genome sequence data from 517 FM chickens representing 44 chicken breeds worldwide, all with the FM phenotype (Fig. 2A). Our comprehensive population
genomic analysis showed that all these breeds have the same structural variations (SVs) at the _FM_ locus as previously reported2. Our population genomic analysis provides overwhelming
support for the FM-2 model based on the pattern of linkage disequilibrium across the complex rearrangement reflecting where recombination events can occur. This study highlights how
population genomics data can be used to resolve complex structural variations that are challenging to resolve even using long-read sequence data. RESULTS COMPREHENSIVE GENOMIC SURVEY OF THE
_FM_ LOCUS We analysed whole genome sequencing data of 517 FM chickens representing 44 breeds of chicken (Fig. 2A). Our results showed that all these breeds have the same structural variants
(SVs) at the _FM_ locus (Fig. 2B and C). The SVs involve duplications of two genomic regions on chromosome 20:10,766,772–10,894,151 bp (DUP1) and chr20:11,306,686–11,477,501 bp (DUP2) (Fig.
2C), consistent with previous findings2,12. GENETIC PROFILE OF THE _FM_ ALLELE We performed a comprehensive analysis of genomic data to characterize the arrangement of the complex
chromosomal structural variation of the _FM_ allele (Figs. 3 and 4). Firstly, we analyzed the sequence depth at the two duplications associated with the FM phenotype and confirmed that DUP1
and DUP2 occur as two copies in the _FM_ allele (Fig. 3A and B). This analysis allowed us to identify _FM_ homozygotes that were used in the further analysis. We next determined the genetic
differentiation between wild-type (_FM*N_/_N_) and _FM_ homozygous chickens (_FM*FM_/_FM_) (Fig. 3C). _F_ST results showed that there were significant genetic differences in the two SV
regions, especially in DUP1 and its flanks. Notably, there is an obvious reduction of genetic differentiation at the front segment of the DUP1 region (Fig. 3C). Nucleotide diversity analysis
showed a marked reduction of heterozygosity in _FM_ homozygotes but interestingly only in the DUP1 region and its flanks (Fig. 3D). We next analyzed the frequency of the reference allele
for all SNPs across the region harboring the two duplications. This revealed a remarkable bubble for the DUP1 region in _FM_ homozygotes not present in wild-type chicken (Fig. 4A and B). In
the bubble region, _FM_ homozygotes tended to be fixed for the reference allele or the non-reference allele, or the two alleles occur at exactly the same frequency (50%) (Fig. 4B). A weak
tendency for a similar pattern was noted for the DUP2 region where two regions (marked with red arrows) had an excess of SNPs showing an allele frequency of 50% (Fig. 4B). The non-duplicated
412.54 kb region between DUP1 and DUP2 showed a very similar distribution of allele frequencies as present in wild-type chickens (Fig. 4A and B). These data are consistent with strong
suppression of recombination between FM and wild-type chromosomes but only for the DUP1 region. CHARACTERIZATION OF THE STRUCTURAL ARRANGEMENT OF _FM_ ALLELE The above results refute FM-1 as
a possible organization of the _FM_ allele. This is because the FM-1 configuration (Fig. 1A) implies an inversion involving one copy of DUP1, the connecting region (412.54 kb) and one copy
of DUP2, and the bubble should extend across the entire region which it does not (Fig. 4B). We therefore conclude that the FM-2 configuration must be the correct organization because it is
the only one consistent with a single previously reported recombination event2 and none of the recent reports based on PacBio long reads found support for the FM-3 order10,11. In addition,
if the configuration of FM-3 is correct, we should observe a bubble in the DUP2 region, not in the DUP1 region. Phylogenetic trees constructed for the DUP1 and DUP2 regions revealed
differences in branch lengths between two SV regions which support the hypothesis of an inversion near the DUP1 region (Supplementary Fig. 1), consistent with our proposed structural
configuration of the _FM_ locus. The large number of SNPs showing an allele frequency of 50% in the DUP1 region implies that the complex rearrangement creating the _FM_ allele must have
involved an inter-chromosomal event involving two different haplotypes creating fixed heterozygosity for the positions in regions of severely suppressed recombination (after the
rearrangement) where the two haplotypes differed (Fig. 4C). In conclusion, FM-2 (Fig. 1A) is now the confirmed organization of the _FM_ allele and there is strong suppression of
recombination for most of the DUP1-inverted DUP2-DUP1 region whereas there is no strong suppression of recombination from the end of the second copy of the DUP1 region to the second copy of
DUP2 including the single copy 412.54 kb intervening region (Fig. 4C). This explains the bubble pattern across the DUP1 region (Fig. 4B) in _FM_ homozygotes as well as the corresponding
pattern of genetic differentiation and nucleotide diversity (Fig. 3C and D). TRACING THE FORMATION OF THE _FM_ ALLELE There is a sharp disruption of the region of fixed heterozygosity within
DUP1 in the _FM_ allele, marked by a red arrow in Fig. 4B. This could either reflect a recombination with wild-type chromosomes have occurred or a shift from a large region where the two
donor haplotypes forming the _FM_ duplication showed many sequence differences to a region where they happened to be identical. Similarly, two reference-consistent regions in DUP2 may
reflect regions in which one of the copies of DUP2 not undergoing recombination is identical to the genome reference and thus the frequency of the reference allele among chickens homozygous
for the _FM_ allele will always be 50% or higher (Fig. 4B), because we estimate the allele frequency as the average of sequences from the two copies of DUP2. Linkage disequilibrium (LD)
analysis at the _FM_ locus also showed a clear LD block boundary between two segments within DUP1 which is in perfect agreement with the break of fixed heterozygosity (Figs. 4B and 5). While
LD decay patterns are influenced by recombination rates, population structure, and historical selection events, the significantly elevated LD at the _FM_ locus (Fig. 5) are consistent with
the presence of an inversion. DISCUSSION RESOLVING THE STRUCTURAL CONFIGURATION OF THE _FM_ ALLELE The _FM_ allele constitutes a truly complex structural variant involving two large
duplications (127.4 kb and 170.5 kb), an inversion and translocation of the 170.5 duplication and with a 412.5 kb non-duplicated intervening region (Fig. 1). Previous PCR studies established
three possible configurations of this complex rearrangement (Fig. 1) that could not be resolved using whole-genome sequencing based on short read data2,12. However, one recombinant found in
a pedigree analysis provided strong support for the FM-2 configuration5. Subsequent studies based on long-read sequencing gave conflicting results, some supporting the FM-1
configuration10,11 and others supporting FM-28,9,13. The current study based on extensive whole genome sequence data and the analysis of linkage disequilibrium patterns across more than 40
breeds carrying the _FM_ allele now provides conclusive evidence for the FM-2 configuration and that the _FM_ allele arose only one time. The unique feature with an inverted copy of DUP2
inserted between two tandem copies of DUP1 results in strong suppression of recombination explaining the allele frequency bubble at DUP1 when reads from the _FM_ allele are aligned to the
reference genome (Fig. 4). Furthermore, the data shows that the _FM_ mutation must have involved an inter-chromosomal exchange resulting in fixed heterozygosity in the DUP1 region at the
sites with sequence differences between the two copies (Fig. 4B). In contrast, fixed heterozygosity and an allele frequency bubble is not noted for DUP2 because one of the DUP2 copies is
located outside the region of suppressed recombination (see FM-2 in Fig. 1A). This result is also in complete agreement with the previously reported recombination event between a wild-type
and an _FM_ allele2. Given that the _FM_ allele likely originated thousands of years ago5, sufficient time has passed for sequence variation in the second copy of DUP2 and the corresponding
region of wild-type chromosomes to become randomized due to recombination. This is illustrated by the fact that the nucleotide diversity is clearly reduced only at the DUP1 region and its
flanking region (Fig. 3D) consistent with the observed allele frequency bubble (Fig. 4). EVOLUTION OF THE _FM_ LOCUS Our study did not reveal any genetic heterogeneity at the _FM_ locus
since exactly the same structural rearrangements were detected across all 44 FM chicken breeds analyzed. The _FM_ allele must involve multiple structural rearrangements and it is challenging
to establish the exact mechanism causing these rearrangements with confidence. However, one possible scenario is that the first event involved a duplication of the entire region from DUP1
to DUP2 (710.4 kb) by unequal cross-over, followed by an inversion involving DUP2 and the intervening sequence (412.5 kb) in which the latter was lost. This scenario is consistent with the
fixed heterozygosity for the DUP1 sequence, because the two copies originate from different chromosome homologs. The fixed heterozygosity at DUP1 has since then been maintained due to
suppressed recombination while the fixed heterozygosity for DUP2 has been eroded due to recombination affecting the second copy of DUP2 (Fig. 4). It is still an open question whether the two
events (the duplication and the inversion) occurred in a single meiosis or in a stepwise fashion over multiple generations. LIMITED CAPABILITY OF LONG-READ DATA IN COMPLEX STRUCTURAL
VARIATION For complex structural variations, such as the _FM_ locus, which involve copy number variation, rearrangements, inversion, and translocations, long-read sequencing faces
limitations for instance when the size of a duplication exceeds the read length. In such cases, the inter-sequence assembly strategy plays a critical role and can significantly influence the
accuracy of the assembly14. As more and more research focus on large-scale structural variation15, such as pan-genome16, we suggest that the assembly of regions showing complex structural
variation can be validated using population genomics data. Despite previous conflicting reports2,10,11,12, the population genomic approach employed here successfully clarified the structural
arrangement, demonstrating the value of population-scale sequencing in deciphering complex genomic rearrangements. STRUCTURAL VARIANTS AS DRIVERS OF PHENOTYPIC DIVERSITY The _FM_ locus
exemplifies how structural variants contribute to phenotypic diversity in domesticated animals17,18,19,20,21,22. These are often gain-of-function mutations with a dominant inheritance,
resulting in altered gene expression. Other prominent examples include Dominant white color in pigs21, Greying with age in horses17,18, and all three comb phenotypes in chicken, Pea-comb23,
Rose-comb24, and Duplex comb22. These cases collectively highlight the evolutionary significance of structural variants, particularly in shaping key morphological traits under artificial
selection in domestic animals. MATERIALS AND METHODS SAMPLES COLLECTION In this study, we analyzed 817 chicken samples, comprising 517 FM chicken samples from 44 breeds from various
geographic locations worldwide, and an additional 300 wild-type samples (Supplementary Table 1). All samples included in this analysis were previously reported in
studies9,10,11,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50. DATA PROCESSING Sequencing reads underwent quality control using FastQC (version 0.11.8) to
assess read quality metrics. Index adaptors and raw reads with more than 50% bases in low quality (Q ≤ 5) or 10% “N” content were filtered out using Btrim (version 0.3.0)51 software. Cleaned
reads were then mapped to the reference genome of the red junglefowl (GRCg6a) using the BWA-MEM (version 0.7.17-r1188)52 algorithm with default settings. Bam files were sorted using
SAMtools (version 1.9)53 and PCR duplicates were removed using Picard (version 2.18.6) tools. Sequencing depth relative to the reference genome was calculated using SAMtools (version 1.9)53.
The “RealignerTargetCreator” and “IndelRealigner” tools in Genome Analysis Toolkit (GATK, version 3.7)54 were used to reduce mismatches around INDELs, with base quality score recalibration
(BQSR) performed to reduce mapping errors. Joint calling for SNPs and small INDELs was performed using the “HaplotypeCaller” and “GenotypeGVCFs” tools from GATK (version 3.7)54. Low quality
SNPs were filtered, with parameters following previous research7,55: “QUAL < 30.0 || QD < 2.0 || MQ < 40.0 || FS > 60.0 || MQRankSum < − 12.5 || ReadPosRankSum < − 8.0 ||
SOR > 3.0”. Loci with max-missing rate above 0.10 and minor allele frequency less than 0.05 (–maf 0.05; –max-missing 0.9) were filtered using VCFtools (version 0.1.16)56. Only biallelic
loci were retained. SVS IDENTIFICATION SVs were detected using relative sequence depth analysis. Sequence depth was calculated using SAMtools (version 1.9)53 to identify the genomic regions
of interest, specifically focusing on the DUP1 (chr20:10,766,772–10,894,151) and DUP2 (chr20:11,306,686–11,477,501) regions. The relative sequencing depth is the ratio of the sequencing
depth of the candidate region to the whole genome background. To differentiate between wild type (_FM*N_/_N_), homozygous (_FM*FM_/_FM_), and heterozygous (_FM*FM_/_N_) samples, relative
sequence depth analysis was performed using the average sequence depth of the target region divided by the average sequence depth of whole genome or chromosome 20. NUCLEOTIDE DIVERSITY AND
GENETIC DIFFERENTIATION Nucleotide diversity (π) and genetic differentiation (_F_ST) was calculated using VCFtools (version 0.1.16)56 using a 20 kb window length with 10 kb sliding window,
focusing on the SV regions and their flanking sequences (chr20:10,600,000–11,700,000). To evaluate the statistical significance of _F_ST estimates across different genomic regions, we
performed a _Z_-score transformation relative to the genome-wide distribution. _P_-values were derived from the standard normal distribution, and Benjamini–Hochberg false discovery rate
(FDR) correction was applied to control for multiple testing. _F_ST windows with FDR-adjusted _P_-values < 0.05 were considered significantly different from the genome-wide expectation.
Allele frequency distributions were analyzed using VCFtools (version 0.1.16)56, with particular attention to differences between wild type, heterozygous, and homozygous FM chickens. Regions
showing significant deviations in allele frequency were identified and further examined for recombination breakpoints manually. PHYLOGENETIC ANALYSIS Phylogenetic relationships among FM
chickens were inferred using the identified SV regions. Neighbor-joining (NJ) trees were constructed using and RapidNJ software and visualized using iTOL57. Separate trees were constructed
for the DUP1 and DUP2 regions. LINKAGE DISEQUILIBRIUM ANALYSIS To assess LD decay patterns across the FM locus, we computed pairwise linkage disequilibrium (LD) using r2 between SNPs using
LDBlockShow (version 1.40)58 with default parameters. LD decay patterns were used to infer historical recombination rates, assuming a constant recombination landscape over time. The LD block
structure was analyzed to identify boundaries within the DUP1 and DUP2 regions, indicating possible recombination breakpoints. To determine whether LD decay in the _FM_ locus was
significantly different from the genome-wide background, we performed a genome-wide _Z_-score transformation within 10 kb windows. We then converted _Z_-scores to _P_-values using the
standard normal distribution and applied FDR correction to control for multiple testing. Regions with FDR-adjusted _P_-values < 0.05 were considered significantly different from
genome-wide LD expectations. This analysis assumes that LD patterns reflect both historical recombination events and population demographic history. Specifically, LD tends to persist in
regions under strong selection, low recombination, or with recent population bottlenecks. DATA AVAILABILITY All of the whole genome sequencing (WGS) datasets used in this study have been
previously reported (Supplementary Table 1). The variant call format (VCF) data was deposited in the Genome Variation Map (GVM) under accession project number PRJCA032154. CODE AVAILABILITY
All analyses were performed using previously published or developed tools, as indicated in Methods. No custom code was developed or used. REFERENCES * Andersson, L. & Purugganan, M.
Molecular genetic variation of animals and plants under domestication. _Proc. Natl. Acad. Sci. USA_ 119, e2122150119. https://doi.org/10.1073/pnas.2122150119 (2022). Article PubMed PubMed
Central CAS Google Scholar * Dorshorst, B. et al. A complex genomic rearrangement involving the endothelin 3 locus causes dermal hyperpigmentation in the chicken. _PLoS Genet._ 7,
e1002412. https://doi.org/10.1371/journal.pgen.1002412 (2011). Article PubMed PubMed Central CAS Google Scholar * Siddiqui, S. A., Toppi, V. & Syiffah, L. A comparative review on
Ayam Cemani chicken - A comparison with the most common chicken species in terms of nutritional values, LCA, price and consumer acceptance. _Trop. Anim. Health Prod._ 56, 161.
https://doi.org/10.1007/s11250-024-03980-6 (2024). Article PubMed PubMed Central CAS Google Scholar * Shoenfeld, N., Amital, H. & Shoenfeld, Y. The effect of melanism and vitamin D
synthesis on the incidence of autoimmune disease. _Nat. Clin. Pract. Rheumatol._ 5, 99–105. https://doi.org/10.1038/ncprheum0989 (2009). Article PubMed MATH CAS Google Scholar * Li, S.
_Compendium of materia medica_ 1st edn, 255 (Overseas Chinese Publishing House, 2017). MATH Google Scholar * Kim, I. S. et al. Microenvironment-derived factors driving metastatic
plasticity in melanoma. _Nat. Commun._ 8, 14343. https://doi.org/10.1038/ncomms14343 (2017). Article ADS PubMed PubMed Central CAS Google Scholar * Ma, C., Shi, X., Li, X., Zhang, Y.
P. & Peng, M. S. Comprehensive evaluation and guidance of structural variation detection tools in chicken whole genome sequence data. _BMC Genomics_ 25, 970.
https://doi.org/10.1186/s12864-024-10875-1 (2024). Article PubMed PubMed Central MATH CAS Google Scholar * Dharmayanthi, A. B. et al. The origin and evolution of fibromelanosis in
domesticated chickens: Genomic comparison of Indonesian Cemani and Chinese Silkie breeds. _PLoS One_ 12, e0173147. https://doi.org/10.1371/journal.pone.0173147 (2017). Article PubMed
PubMed Central CAS Google Scholar * Shinde, S. S., Sharma, A. & Vijay, N. Decoding the fibromelanosis locus complex chromosomal rearrangement of black-bone chicken: Genetic
differentiation, selective sweeps and protein-coding changes in Kadaknath chicken. _Front. Genet._ 14, 1180658. https://doi.org/10.3389/fgene.2023.1180658 (2023). Article PubMed PubMed
Central CAS Google Scholar * Sohn, J. I. et al. Whole genome and transcriptome maps of the entirely black native Korean chicken breed Yeonsan Ogye. _GigaScience_
https://doi.org/10.1093/gigascience/giy086 (2018). Article PubMed PubMed Central MATH Google Scholar * Zhu, F. et al. A chromosome-level genome assembly for the Silkie chicken resolves
complete sequences for key chicken metabolic, reproductive, and immunity genes. _Commun. Biol._ 6, 1233. https://doi.org/10.1038/s42003-023-05619-y (2023). Article PubMed PubMed Central
MATH CAS Google Scholar * Shinomiya, A. et al. Gene duplication of endothelin 3 is closely correlated with the hyperpigmentation of the internal organs (Fibromelanosis) in silky chickens.
_Genetics_ 190, 627–638. https://doi.org/10.1534/genetics.111.136705 (2012). Article PubMed PubMed Central MATH CAS Google Scholar * Sharma, A. & Vijay, N. What is the correct
genomic structure of the complex chromosomal rearrangement at the Fm locus in Silkie chicken?. _bioRxiv_ https://doi.org/10.1101/2024.02.05.578760 (2024). Article PubMed PubMed Central
MATH Google Scholar * Ahsan, M. U., Liu, Q., Perdomo, J. E., Fang, L. & Wang, K. A survey of algorithms for the detection of genomic structural variants from long-read sequencing data.
_Nat. Methods_ 20, 1143–1158. https://doi.org/10.1038/s41592-023-01932-w (2023). Article PubMed PubMed Central MATH CAS Google Scholar * Ho, S. S., Urban, A. E. & Mills, R. E.
Structural variation in the sequencing era. _Nat. Rev. Genet._ 21, 171–189. https://doi.org/10.1038/s41576-019-0180-9 (2020). Article PubMed MATH CAS Google Scholar * Liao, W. W. et al.
A draft human pangenome reference. _Nature_ 617, 312–324. https://doi.org/10.1038/s41586-023-05896-x (2023). Article ADS PubMed PubMed Central MATH CAS Google Scholar * Rubin, C. J.
et al. An intronic copy number variation in Syntaxin 17 determines speed of greying and melanoma incidence in Grey horses. _Nat. Commun._ 15, 7510. https://doi.org/10.1038/s41467-024-51898-2
(2024). Article PubMed PubMed Central MATH CAS Google Scholar * Rosengren Pielberg, G. et al. A cis-acting regulatory mutation causes premature hair graying and susceptibility to
melanoma in the horse. _Nat. Genet._ 40, 1004–1009. https://doi.org/10.1038/ng.185 (2008). Article PubMed CAS Google Scholar * Liu, X. et al. Evolutionary origin of genomic structural
variations in domestic yaks. _Nat. Commun._ 14, 5617. https://doi.org/10.1038/s41467-023-41220-x (2023). Article ADS PubMed PubMed Central MATH CAS Google Scholar * Serres-Armero, A.
et al. Copy number variation underlies complex phenotypes in domestic dog breeds and other canids. _Genome Res._ 31, 762–774. https://doi.org/10.1101/gr.266049.120 (2021). Article PubMed
PubMed Central CAS Google Scholar * Giuffra, E. et al. A large duplication associated with dominant white color in pigs originated by homologous recombination between LINE elements
flanking KIT. _Mamm. Genome_ 13, 569–577. https://doi.org/10.1007/s00335-002-2184-5 (2002). Article PubMed CAS Google Scholar * Dorshorst, B. et al. A genomic duplication is associated
with ectopic eomesodermin expression in the embryonic chicken comb and two duplex-comb phenotypes. _PLoS Genet._ 11, e1004947. https://doi.org/10.1371/journal.pgen.1004947 (2015). Article
PubMed PubMed Central CAS Google Scholar * Wright, D. et al. Copy number variation in intron 1 of SOX5 causes the Pea-comb phenotype in chickens. _PLoS Genet._ 5, e1000512.
https://doi.org/10.1371/journal.pgen.1000512 (2009). Article PubMed PubMed Central CAS Google Scholar * Imsland, F. et al. The Rose-comb mutation in chickens constitutes a structural
rearrangement causing both altered comb morphology and defective sperm motility. _PLoS Genet._ 8, e1002775. https://doi.org/10.1371/journal.pgen.1002775 (2012). Article PubMed PubMed
Central MATH CAS Google Scholar * Ulfah, M. et al. Genetic features of red and green junglefowls and relationship with Indonesian native chickens Sumatera and Kedu Hitam. _BMC Genomics_
17, 320. https://doi.org/10.1186/s12864-016-2652-z (2016). Article PubMed PubMed Central CAS Google Scholar * Wang, M. S. et al. 863 genomes reveal the origin and domestication of
chicken. _Cell Res._ 30, 693–701. https://doi.org/10.1038/s41422-020-0349-y (2020). Article PubMed PubMed Central MATH CAS Google Scholar * Youm, D. J. et al. The idiosyncratic genome
of Korean long-tailed chicken as a valuable genetic resource. _iScience_ 26, 106236. https://doi.org/10.1016/j.isci.2023.106236 (2023). Article ADS PubMed PubMed Central MATH CAS
Google Scholar * Chen, X. et al. Population genomic sequencing delineates global landscape of copy number variations that drive domestication and breed formation of in chicken. _Front.
Genet._ 13, 830393. https://doi.org/10.3389/fgene.2022.830393 (2022). Article PubMed PubMed Central CAS Google Scholar * Wang, Y. M. et al. Integrating genomic and transcriptomic data
to reveal genetic mechanisms underlying piao chicken rumpless trait. _Genomics Proteomics Bioinform._ 19, 787–799. https://doi.org/10.1016/j.gpb.2020.06.019 (2021). Article MATH Google
Scholar * Zhi, Y. et al. Genome-wide genetic structure of Henan indigenous chicken breeds. _Animals_ https://doi.org/10.3390/ani13040753 (2023). Article PubMed PubMed Central MATH
Google Scholar * Beauclair, L. et al. Sequence properties of certain GC rich avian genes, their origins and absence from genome assemblies: case studies. _BMC Genomics_ 20, 734.
https://doi.org/10.1186/s12864-019-6131-1 (2019). Article PubMed PubMed Central MATH CAS Google Scholar * Bortoluzzi, C. et al. Parallel genetic origin of foot feathering in birds.
_Mol. Biol. Evolut._ 37, 2465–2476. https://doi.org/10.1093/molbev/msaa092 (2020). Article MATH CAS Google Scholar * Fan, W. L. et al. Genome-wide patterns of genetic variation in two
domestic chickens. _Genome Biol. Evolut._ 5, 1376–1392. https://doi.org/10.1093/gbe/evt097 (2013). Article MATH Google Scholar * Yi, G. et al. Genome-wide patterns of copy number
variation in the diversified chicken genomes using next-generation sequencing. _BMC Genomics_ 15, 962. https://doi.org/10.1186/1471-2164-15-962 (2014). Article PubMed PubMed Central MATH
Google Scholar * Noorai, R. E., Shankar, V., Freese, N. H., Gregorski, C. M. & Chapman, S. C. Discovery of genomic variations by whole-genome resequencing of the North American
Araucana chicken. _PLoS One_ 14, e0225834. https://doi.org/10.1371/journal.pone.0225834 (2019). Article PubMed PubMed Central CAS Google Scholar * Chen, C. et al. Whole-genome
resequencing reveals melanin deposition candidate genes of Luning chicken. _BMC Genomics_ 25, 858. https://doi.org/10.1186/s12864-024-10774-5 (2024). Article PubMed PubMed Central MATH
CAS Google Scholar * Gu, J., Liang, Q., Liu, C. & Li, S. Genomic analyses reveal adaptation to hot arid and harsh environments in native chickens of China. _Front. Genet._ 11, 582355.
https://doi.org/10.3389/fgene.2020.582355 (2020). Article PubMed PubMed Central CAS Google Scholar * Lawal, R. A. et al. The wild species genome ancestry of domestic chickens. _BMC
Biol._ 18, 13. https://doi.org/10.1186/s12915-020-0738-1 (2020). Article PubMed PubMed Central MATH CAS Google Scholar * Tian, S. et al. Genomic analyses reveal genetic adaptations to
tropical climates in chickens. _iScience_ 23, 101644. https://doi.org/10.1016/j.isci.2020.101644 (2020). Article ADS PubMed PubMed Central CAS Google Scholar * Zhao, X. et al.
Significant genomic introgression from grey junglefowl (_Gallus sonneratii_) to domestic chickens (_Gallus gallus domesticus_). _J. Anim. Sci. Biotechnol._ 15, 45.
https://doi.org/10.1186/s40104-024-01006-7 (2024). Article PubMed PubMed Central CAS Google Scholar * Chu, J. et al. The genomic characteristics affect phenotypic diversity from the
perspective of genetic improvement of economic traits. _iScience_ 26, 106426. https://doi.org/10.1016/j.isci.2023.106426 (2023). Article ADS PubMed PubMed Central MATH CAS Google
Scholar * Li, M. et al. De novo assembly of 20 chicken genomes reveals the undetectable phenomenon for thousands of core genes on microchromosomes and subtelomeric regions. _Mol. Biol.
Evolut._ https://doi.org/10.1093/molbev/msac066 (2022). Article Google Scholar * Guo, Y. et al. Researching on the fine structure and admixture of the worldwide chicken population reveal
connections between populations and important events in breeding history. _Evolut. Appl._ 15, 553–564. https://doi.org/10.1111/eva.13241 (2022). Article MATH Google Scholar * Wu, M. Y. et
al. Historic and modern genomes unveil a domestic introgression gradient in a wild red junglefowl population. _Evolut. Appl._ 13, 2300–2315. https://doi.org/10.1111/eva.13023 (2020).
Article MATH Google Scholar * Li, D. et al. Breeding history and candidate genes responsible for black skin of Xichuan black-bone chicken. _BMC Genomics_ 21, 511.
https://doi.org/10.1186/s12864-020-06900-8 (2020). Article PubMed PubMed Central MATH CAS Google Scholar * Zeng, T. et al. Analysis of genome and methylation changes in Chinese
indigenous chickens over time provides insight into species conservation. _Commun. Biol._ 5, 952. https://doi.org/10.1038/s42003-022-03907-7 (2022). Article PubMed PubMed Central MATH
CAS Google Scholar * Zhong, H. A. et al. Microevolutionary mechanism of high-altitude adaptation in Tibetan chicken populations from an elevation gradient. _Evolut. Appl._ 15, 2100–2112.
https://doi.org/10.1111/eva.13503 (2022). Article MATH CAS Google Scholar * Tan, X. et al. Whole-genome variants dataset of 209 local chickens from China. _Sci. Data_ 11, 169.
https://doi.org/10.1038/s41597-024-02995-w (2024). Article PubMed PubMed Central MATH CAS Google Scholar * Xu, D. et al. Whole-genome sequencing revealed genetic diversity, structure
and patterns of selection in Guizhou indigenous chickens. _BMC Genomics_ 24, 570. https://doi.org/10.1186/s12864-023-09621-w (2023). Article PubMed PubMed Central MATH CAS Google
Scholar * Bendesky, A. et al. The main genetic locus associated with the evolution of gamecocks is centered on ISPD. _G3-Genes Genomes Genet._ https://doi.org/10.1093/g3journal/jkad267
(2024). Article Google Scholar * Kong, Y. Btrim: A fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. _Genomics_ 98, 152–153.
https://doi.org/10.1016/j.ygeno.2011.05.009 (2011). Article PubMed MATH CAS Google Scholar * Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler
transform. _Bioinformatics_ 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009). Article PubMed PubMed Central MATH CAS Google Scholar * Li, H. et al. The sequence
alignment/map format and SAMtools. _Bioinformatics_ 25, 2078–2079. https://doi.org/10.1093/bioinformatics/btp352 (2009). Article PubMed PubMed Central MATH CAS Google Scholar *
McKenna, A. et al. The genome analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. _Genome Res._ 20, 1297–1303. https://doi.org/10.1101/gr.107524.110
(2010). Article PubMed PubMed Central MATH CAS Google Scholar * Gu, L. H. et al. Genomic insights into local adaptation and phenotypic diversity of Wenchang chickens. _Poultry Sci._
103, 103376. https://doi.org/10.1016/j.psj.2023.103376 (2024). Article MATH CAS Google Scholar * Danecek, P. et al. The variant call format and VCFtools. _Bioinformatics_ 27, 2156–2158.
https://doi.org/10.1093/bioinformatics/btr330 (2011). Article PubMed PubMed Central MATH CAS Google Scholar * Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v6: Recent
updates to the phylogenetic tree display and annotation tool. _Nucleic Acids Res._ 52, W78–W82. https://doi.org/10.1093/nar/gkae268 (2024). Article PubMed PubMed Central Google Scholar *
Dong, S. S. et al. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. _Brief. Bioinform._
https://doi.org/10.1093/bib/bbaa227 (2021). Article PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank Min-Sheng Peng (Kunming Institute of Zoology,
Chinese Academy of Sciences, Kunming, China) and Xun-He Huang (School of Life Science, Jiaying University, Meizhou, China) for their support and discussion. FUNDING Open access funding
provided by Uppsala University. The project was financially supported by Vetenskapsrådet (2017-02907), Knut and Alice Wallenberg Foundation (KAW 2023.0160), and Erik Philip-Sörensen
Foundation (G2024-033). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Medical Biochemistry and Microbiology, Uppsala University, Uppsala, Sweden Cheng Ma & Leif Andersson *
Department of Veterinary Integrative Biosciences, Texas A&M University, College Station, USA Leif Andersson Authors * Cheng Ma View author publications You can also search for this
author inPubMed Google Scholar * Leif Andersson View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS C.M. and L.A conceived and designed the
project. C.M. performed bioinformatic analyses. C.M. and L.A. wrote the manuscript. CORRESPONDING AUTHORS Correspondence to Cheng Ma or Leif Andersson. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. ETHICAL APPROVAL Data were directly downloaded from published studies and no additional ethics approval was needed. Each study is
referenced and details on ethics approval are available in each manuscript. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION 1. SUPPLEMENTARY INFORMATION 2. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed
under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ma, C., Andersson, L. Population genomic analysis
identifies the complex structural variation at the fibromelanosis (_FM_) locus in chicken. _Sci Rep_ 15, 9239 (2025). https://doi.org/10.1038/s41598-025-94250-4 Download citation * Received:
04 December 2024 * Accepted: 12 March 2025 * Published: 18 March 2025 * DOI: https://doi.org/10.1038/s41598-025-94250-4 SHARE THIS ARTICLE Anyone you share the following link with will be
able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing
initiative