Unraveling the independent role of mettl3 in m6a modification and tumor progression in esophageal squamous cell carcinoma
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT METTL3 and METTL14 are traditionally posited to assemble the m6A methyltransferase complex in a stoichiometric 1:1 ratio, modulating mRNA fate via m6A modifications. Nevertheless,
recent investigations reveal inconsistent expression levels and prognostic significance of METTL3 and METTL14 across various tumor types, challenging their consistent functional engagement
in neoplastic contexts. A pan-cancer analysis leveraging The Cancer Genome Atlas (TCGA) data has identified pronounced disparities in the expression patterns, functional roles, and
correlations with tumor burden between METTL3 and METTL14, particularly in esophageal squamous cell carcinoma (ESCC). Knockdown experiments of METTL3 in EC109 cells markedly suppress cell
proliferation both in vitro and in vivo, whereas METTL14 knockdown shows a comparatively muted effect on proliferation and does not significantly alter METTL3 protein levels. mRNA sequencing
indicates that METTL3 singularly governs the expression of 1615 genes, with only 776 genes co-regulated with METTL14. Additionally, immunofluorescence co-localization studies suggest
discrepancies in cellular localization between METTL3 and METTL14. High-performance liquid chromatography–mass spectrometry (HPLC–MS) analyses demonstrate that METTL3 uniquely associates
with the Nop56p-linked pre-rRNA complex and mRNA splicing machinery, independent of METTL14. Preliminary bioinformatics and multi-omics investigations reveal that METTL3’s autonomous role in
modulating tumor cell proliferation and its involvement in mRNA splicing are potentially pivotal molecular mechanisms. Our study lays both experimental and theoretical groundwork for a
deeper understanding of the m6A methyltransferase complex and the development of targeted tumor therapies focusing on METTL3. SIMILAR CONTENT BEING VIEWED BY OTHERS MULTI-OMICS INTEGRATION
OF METHYLTRANSFERASE-LIKE PROTEIN FAMILY REVEALS CLINICAL OUTCOMES AND FUNCTIONAL SIGNATURES IN HUMAN CANCER Article Open access 20 July 2021 NSUN2-MEDIATED RNA 5-METHYLCYTOSINE PROMOTES
ESOPHAGEAL SQUAMOUS CELL CARCINOMA PROGRESSION VIA LIN28B-DEPENDENT GRB2 MRNA STABILIZATION Article Open access 03 August 2021 METTL3 PROMOTES TUMOUR DEVELOPMENT BY DECREASING APC EXPRESSION
MEDIATED BY _APC_ MRNA _N_6-METHYLADENOSINE-DEPENDENT YTHDF BINDING Article Open access 21 June 2021 INTRODUCTION METTL3 serves as the catalytic subunit of the N6-adenosine
methyltransferase complex and, in conjunction with METTL14, comprises the “writer” complex essential for catalyzing m6A modifications on mRNA1. The presence of m6A modifications on mRNA
facilitates their recognition by “reader” proteins, thereby enhancing the processes of mRNA translation or degradation2. m6A modifications can also be reversed by “eraser” enzymes. The
METTL3–METTL14-mediated m6A modification plays a pivotal role in regulating cellular proliferation, differentiation, and responses to various stresses. In recent years, mRNA m6A methylation
mediated by the METTL3–METTL14 complex has offered a novel paradigm for elucidating the mechanisms underlying cancer progression. Numerous studies have demonstrated that the upregulation of
METTL3, frequently observed in tumors, promotes tumor cell survival, proliferation, self-renewal, metastasis, and drug resistance by modulating mRNA metabolism3. Elevated expression of
METTL3/METTL14 has been significantly correlated with the advancement and adverse prognosis of various cancers4. In acute myeloid leukemia, the overexpression of METTL3/METTL14 enhances the
stability and translation efficiency of c-myc via m6A modifications, thereby accelerating cancer progression5. In breast cancer, the METTL3-METTL14 complex upregulates the expression of
genes like Bcl-2 and CXCR4 through m6A-dependent mechanisms, thereby fostering breast cancer growth and metastasis6. Nevertheless, in certain cancers, METTL3 and METTL14 demonstrate
divergent effects on tumor progression. Knockdown of METTL3 has been observed to alter the malignant phenotype across various tumor types, whereas multiple studies have indicated that
elevated expression of METTL14 inhibits tumor proliferation and migration via m6A-dependent mechanisms. These contrasting prognostic impacts have been documented in colorectal, bladder,
lung, gastric, and other cancers7,8. Additional research has demonstrated that the regulatory effect of METTL3 on downstream genes may operate independently of METTL14. For instance, in
ovarian cancer, METTL3 shows a significant association with prognosis; however, the knockdown of METTL14 does not inhibit the clonogenic formation capacity of TOV-112D cells. Moreover, the
expression of oncogenes such as EIF3C, AXL, CSF1, and FZD10 is solely modulated by METTL3, underscoring its unique regulatory role9. Similarly, in esophageal squamous cell carcinoma (ESCC),
METTL3 and METTL14 demonstrate distinct functional roles. Specifically, the modulation of miR-99a-5p by METTL14 occurs independently of METTL3, highlighting differential regulatory pathways
within this cancer type10,11. The findings from the referenced studies elucidate that while METTL3 and METTL14, integral components of the m6A writer complex, collaboratively influence m6A
modifications in cells, they display marked disparities in their influence on downstream gene regulation in tumor cells, either directly or indirectly. The distinct mechanisms by which
METTL14 regulates genes independently of METTL3 pose critical questions for future m6A research. In our current study, employing a pan-cancer analytical approach, we developed METTL3 and
METTL14 knockdown EC109 esophageal cancer cell lines to explore the impact of gene silencing on cellular proliferation. Additionally, comprehensive transcriptomic and proteomic analyses were
conducted to delineate the intricate molecular mechanisms involved. MATERIALS AND METHODS PAN-CANCER ANALYSIS We retrieved RNA-sequencing expression profiles at level 3 and associated
clinical data for different cancer types from the Cancer Genome Atlas (TCGA) database, accessible via the website https://portal.gdc.com. Subsequent statistical analysis was carried out
using R version 4.0.3 software, provided by the R Foundation for Statistical Computing, based in Vienna, Austria, with a significance threshold set at P-value < 0.05. All the analyses and
implementations of the aforementioned methods were conducted using R version 4.0.3, which is supported by the R Foundation for Statistical Computing (2020). We utilized the ggplot2 package
for creating clear and concise graphics, and the pheatmap package for generating informative heatmaps to visualize our data. CORRELATION MAP We applied Spearman’s correlation analysis to
quantify the relationship between non-normally distributed quantitative variables. For determining statistical significance, we adopted a P-value cutoff of less than 0.05, with results
achieving P < 0.05 being considered statistically noteworthy. PATHWAYS CLUSTERING The clustering analysis were performed at Metascape (https://metascape.org/)12. TRANSFECTION The EC109
cells utilized in the experiments were obtained from the Cell Resource Center, Peking Union Medical College (PCRC). The cells were cultured in high-glucose DMEM medium, incubated at 37 °C in
a 5% CO2 incubator, with the medium containing 10% serum. Lentiviral vector targeting METTL3 and METTL14 were purchased from Bocui technology (Shanxi China). The plasmid
pLKO.1-U6-EF1a-mcherry(copGFP)-T2A-Neo(puro) were was constructed to knockdown the expression of the target gene. After virus infection, G418 or puromycin is used for selection. The sh-RNA
sequence used for knockdown is as follows: * sh-METTL14-1: AAGGATGAGTTAATAGCTAAACTCGAGTTTAGCTATTAACTCATCCTT * sh-METTL14-2: * TGGTGCCGTGTTAAATAGCAACTCGAGTTGCTATTTAACACGGCACCA * sh-METTL3-1:
CTGCAAGTATGTTCACTATGACTCGAGTCATAGTGAACATACTTGCAG * sh-METTL3-2: * AGGAGCCAGCCAAGAAATCAACTCGAGTTGATTTCTTGGCTGGCTCCT WESTERN BLOT To isolate total protein from esophageal carcinoma cells, RIPA
lysis buffer (Beyotime, Shanghai, China) was utilized. Following the extraction process, the membranes were washed six times with TBST for five minutes each. Subsequently, they were
incubated with HRP-conjugated secondary antibodies specific to rabbit or mouse IgG at a dilution of 1:5000, at 4 °C for a 12-h period. Protein levels were detected using the ChemiDOC™ XRS +
imaging system (Bio-Rad). For quantification of protein bands, Olympus Image-Pro Plus software was employed. The immunoblots were captured digitally using said imaging equipment. The
antibodies applied in the western blot analysis included: anti-METTL3 (Abcam, ab195352), anti-METTL14 (Abcam, ab300104), anti-beta-actin (Abcam, ab8226), anti-HNRNPM (Proteintech,
26897-1-AP), anti-U2AF1 (Propteintech, 10334-1-AP), Goat Anti-Rabbit IgG H&L (HRP) (Abcam, ab205718), and Goat Anti-Mouse IgG H&L (HRP) (Abcam, ab205719). Imaging was exclusively
performed using a chemiluminescence module. In Fig. 2E, for which more comprehensive original data cannot be provided. This is due to the section above 50 kDa being excised for use in other
experiments during the experimental process. CLONAL FORMATION ASSAY EC109 cells underwent an initial trypsinization process, followed by cell counting. Then, 1000 cells were seeded into each
well of a 6-well plate. These cells were cultured in a humidified incubator set at 37 °C with 5% CO2 for a duration of 10 days, which allowed for colony formation. After the colonies had
formed, we discarded the culture medium and gently washed the cells with PBS. For fixation, formaldehyde was applied, after which the cells were stained using crystal violet solution (Sangon
Biotech, E607309) to visualize the colonies. TUMOR-BEARING MOUSE MODELS The conduct of mouse-related experiments was approved by the animal ethics committee at Changzhi Medical College,
ensuring adherence to all pertinent guidelines and regulations. We ensured compliance with the ARRIVE guidelines throughout the execution of mouse experiments. All mice were housed in
specific pathogen-free conditions, with regulations on temperature and humidity maintained, alongside a consistent 12-h light/dark cycle. Mice consumed soya-free laboratory chow and had
access to tap water ad libitum. EC109 cells were subcutaneously injected into the left flank of 5-week-old female BALB/c nude mice. Subsequent to the injection, tumor size and survival rate
were monitored bi-daily. Once the tumors attained a volume of 300 mm3, the animals were humanely euthanized through CO2 inhalation in concordance with ethical practices. APOPTOSIS ANALYSIS
Cells undergoing routine culture were digested with EDTA-free trypsin to dissociate them into a single cell suspension after reaching confluence. The detached cells were then suspended in
PBS, and subsequently stained with Annexin V-FITC and propidium iodide (PI) (Beyotime, C1062S) for apoptosis analysis. This staining procedure took place in a dark environment for 20 min to
prevent photobleaching of the fluorescent dyes. Immediately following the incubation period, the stained cells were analyzed using a flow cytometer for apoptosis detection. MRNA-SEQ Total
RNA was extracted employing the Trizol reagent (Thermo Fisher) in strict accordance with the instructions provided by the manufacturer. The isolated RNA was then utilized to construct an
RNA-seq library using the NEBNext® Ultra™ II Directional RNA Library Prep Kit for Illumina (NEB). Sequencing was performed for both non-immunoprecipitated (initial sample) and
m6A-immunoprecipitated RNA (m6A IP sample), involving 150 bp paired-end sequencing on the Illumina HiSeq platform. The sequencing data obtained was of high quality, with a Q30 score
assessment conducted to ensure reliability. Trimming of 3ʹ adaptors and removal of low-quality reads were executed using cutadapt software (version 1.9.3). Cleaned reads were aligned to the
reference human genome (UCSC HG19) with the aid of the Hisat2 software (version 2.0.4). IMMUNOFLUORESCENCE STAINING SCC cells were cultured on coverslips, washed with PBS, and fixed with 4%
paraformaldehyde. Post-fixation, they were blocked with bovine serum albumin for 60 min and incubated with anti-METTL14 and anti-METTL3 antibodies overnight at 4 °C. Following three PBS
washes, cells were incubated for 60 min with Alexa Fluor® 488-conjugated goat anti-rabbit IgG (Abcam, ab150077, 1:1000) and Alexa Fluor® 594-conjugated goat anti-mouse IgG (Abcam, ab150116,
1:1000). After mounting with Sigma’s mounting medium (F6057), cells were observed under an Olympus IX73 microscope. ESCC tissue microarrays were purchased from Shanghai Outdo Biotech in
Shanghai, China (HEsoS060CS01). Experiments using human samples have been approved by the Human Ethics Committee. The immunofluorescence colocalization analysis was conducted using Olympus
analysis software (cellSens Dimension). Initially, pseudocolor was added to the fluorescence images, followed by multichannel fusion. Deconvolution was then performed using the nearest
neighbor algorithm. Finally, the colocalization analysis module was utilized for analysis. LC–MS/MS ANALYSIS The phosphoproteomic study was conducted using a Thermo Fisher Scientific Easy
nLC 1200 system, paired with a Q Exactive HF-X mass spectrometer. Phopeptides were loaded onto a custom-packed column with buffer A and eluted over a 110-min period at a flow rate of 300
nL/min, utilizing a linear increase of buffer B covering 2–40%. Mass spectra were collected in a range from m/z 350 to m/z 1800, at a resolution of 60,000 at m/z 200, and a maximum injection
time of 50 ms per scan. The top 15 most intense precursor ions from the MS scan were selected for higher-energy collisional dissociation (HCD) MS/MS with an isolation window of 1.6 Th,
dynamic exclusion for 30 s, and a resolution of 15,000 at m/z 200. STATISTICS In this study, comparisons between two groups were conducted using Student’s t-test, and comparisons among
multiple groups were performed via one-way ANOVA. The results are presented as mean ± standard deviation. STATEMENT All methods were carried out in accordance with relevant guidelines and
regulations. All experimental protocols were approved by Changzhi medical college licensing committee. We confirm that informed consent was obtained from all subjects and/or their legal
guardians. RESULTS DIFFERENTIAL EXPRESSION AND FUNCTIONAL DISPARITIES OF METTL3 AND METTL14 IN ESCC Initially, we interrogated the TCGA database for prevalent tumors, observing distinct mRNA
expression patterns for METTL3 and METTL14, which typically form a 1:1 complex in healthy tissues. Notably, METTL3 was significantly upregulated across various tumors, especially within the
digestive system (e.g., ESCA, STAD, LIHC, COAD, READ), as depicted in Fig. S1A. In contrast, METTL14’s expression was generally reduced compared to normal tissues, as shown in Fig. S1B.
Interestingly, in some cancers, predictive models incorporating only METTL3 and METTL14, constructed using Cox or Lasso algorithms, effectively generated prognostic signatures, where their
contributions to the risk scores were diametrically opposed (ESCC: Risk score = (0.1783) × METTL3 + (− 0.4982) × METTL14), as illustrated in Fig. S2. Further analysis revealed significant
disparities in tumor mutation burden and microsatellite stability between METTL3 and METTL14, shown in Fig. S3A,B, respectively. Subsequent investigations into ESCA and ESCC confirmed
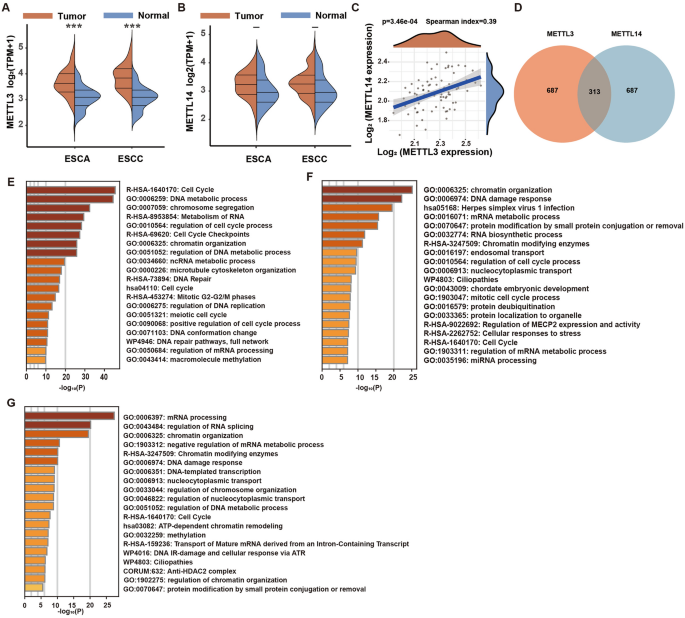
significant alterations in mRNA expression for these genes. In ESCA, METTL3 levels were markedly higher than in normal tissue (Fig. 1A), whereas METTL14 levels did not differ significantly
(Fig. 1B). A correlation study between METTL3 and METTL14 mRNA expressions highlighted a modest positive correlation (Spearman coefficient = 0.39), as detailed in Fig. 1C. Additionally, we
analyzed the top 1000 genes that were positively correlated with each gene. A Venn diagram showed that only 313 of these genes were common between METTL3 and METTL14 (Fig. 1D). Cluster
analysis of METTL3-specific positively correlated genes showed significant enrichment in cell cycle, DNA metabolic processes, and chromosome segregation pathways (Fig. 1E), whereas genes
co-positively correlated with both displayed notable enrichment in chromatin organization, DNA damage, and mRNA metabolism pathways (Fig. 1F). Contrastingly, the enrichment pathways for
METTL14-specific genes included chromatin organization, DNA repair, and mRNA splicing (Fig. 1G). Despite the normal 1:1 ratio formation between METTL3 and METTL14 in healthy cells, gene
mutations, abnormal regulation, or changes in environmental factors during the tumor development process may disrupt this balance, resulting in differences in their expression and function.
KNOCKDOWN OF METTL3 OR METTL14 HAS DIFFERENT EFFECTS ON CELL GROWTH Initially, we effectively knocked down the expression of METTL3 (Fig. 2A,B) and METTL14 (Fig. 2C,D) in EC109 cells using
two distinct siRNAs for each gene. Subsequently, to achieve a more efficient knockdown of METTL3 and METTL14, in the following experiments, we co-transfected the cells with two siRNAs
targeting the same gene; this was done separately in EC109, KYS150, and EC9706 cells to reduce the expression of the target genes. Surprisingly, knockdown of METTL3 significantly reduced the
protein levels of both METTL3 and METTL14, whereas knockdown of METTL14 did not effectively suppress the expression of METTL3 (Fig. 2E–J), with varying degrees of this phenomenon observed
across all three cell lines. Further experiments revealed that in EC109 cells, knockdown of METTL3 significantly inhibited cell proliferation, whereas knockdown of METTL14 had no substantial
impact on the growth and clonogenic ability of EC109 cells (Fig. S4). Flow cytometry analysis showed that cells with METTL3 knockdown exhibited a higher baseline apoptosis rate (Fig. 2K,L).
Additionally, a subcutaneous tumor model in nude mice demonstrated that loss of METTL3 significantly inhibited the tumorigenic ability of EC109 cells, while loss of METTL14 did not
significantly affect tumor growth (Fig. 2M). In summary, our in vitro and in vivo experiments have elucidated the differing roles of METTL3 and METTL14 proteins in tumor cell proliferation
capability. TRANSCRIPTOME SEQUENCING OF METTL3 OR METTL14 KNOCKDOWN CELLS Through mRNA-seq, we observed the impact of METTL3 or METTL14 knockdown on the mRNA expression profile of EC109
cells. After METTL3 knockdown, 2392 genes were downregulated and 396 genes were upregulated, while after METTL14 knockdown, 887 downregulated genes and 132 upregulated genes detected (Table
S1). Venn analysis revealed that among the downregulated genes caused by METTL3 and METTL14 knockdown, 776 genes were co-regulated by both METTL3 and METTL14, 1615 genes were independently
regulated by METTL3, and only 111 genes were independently regulated by METTL14 (Fig. 3A). Cluster analysis showed that METTL3 and METTL14 co-regulated pathways such as chromosome
organization, DNA metabolism, cell division, and Rho GTPases (Fig. 3B). Pathways involved in independently regulated by METTL3 were protein modification by small protein conjugation, DNA
damage response, phosphorylation, regulation of cell cycle process, Golgi vesicle transport, regulation of cellular response to stress, and autophagy etc. (Fig. 3C). The mRNA-seq data
further confirmed that knockdown of METTL3 significantly reduced the expression of METTL14, while knockdown of METTL14 had no effect on the mRNA expression of METTL3 (Fig. 3D,E). PROTEOMIC
IDENTIFICATION BINDING PARTNERS OF METTL3 INDEPENDENT OF METTL14 Based on the aforementioned study, we further hypothesize that METTL3 has independent functions from METTL14 in esophageal
cancer cells (EC109), possibly through the interaction with other complexes. Therefore, we first examined the subcellular localization of METTL3 and METTL14 through co-immunofluorescence
staining in tumor tissues (Fig. 4A,B) and EC109 cells (Fig. 4C,D). The results showed that neither METTL3 nor METTL14 exhibited cytoplasmic localization, and their nuclear localization
exhibited substantial deviation, particularly in tumor tissues. It further reinforced our hypothesis that METTL3 has independent binding partners from METTL14. Subsequently, we performed
co-immunoprecipitation using anti-METTL3 and anti-METTL14 antibodies followed by HPLC–MS identification of the pulled-down proteins. The identification results revealed that METTL3 has
binding partners independent of METTL14, including 56 proteins (Fig. 4E). Furthermore, based on the iBAQ values, we observed that the abundance of METTL14 pulled down by the anti-METTL3
antibody was only about 20% of the abundance of METTL3 (Fig. 4F). Cluster analysis revealed that METTL3’s independent binding partners mainly involved the Nop56p-associated pre-rRNA complex,
mRNA splicing complex, and other signaling pathways such as neutrophil degranulation and protein refolding (Fig. 4G). The protein network analysis was shown in Fig. 4H. We detected
differences in the binding of the splicing factor HNRNPM and U2FA1 protein by METTL3/METTL14 through protein immunoprecipitation (Fig. 4I–J). DISCUSSION In normal cells, METTL3 and METTL14
interact to form a heterodimer with a stoichiometry of 1:1, which facilitates the methylation of RNA via m6A modifications. A pan-cancer analysis leveraging data from TCGA revealed that, in
contrast to normal tissues, mRNA expression levels of METTL3 are significantly upregulated in tumor tissues, whereas the expression levels of METTL14 remain unchanged (Fig. S1). Furthermore,
co-immunoprecipitation studies in tumor cells have demonstrated that the binding affinity of METTL3 for METTL14 is significantly lower than that of METTL14 for METTL3. This disparity
suggests an excess of METTL3 protein abundance relative to METTL14, with the surplus METTL3 not participating in or forming the methyltransferase ‘writer’ complex with METTL1413. In normal
cells, the loss of either METTL3 or METTL14 subunit leads to the degradation of the other subunit14,15. However, in tumor cells, the loss of METTL3 can decrease the protein expression of
METTL14, while knockdown of METTL14 does not significantly affect METTL3 (Fig. 2E–G)16. This indicates that the protein stability of METTL3 and METTL14 is no longer mutually dependent.
METTL3 is significantly upregulated in various tumors and regulates signaling pathways such as p53, Ras/Raf/ERK, and Wnt/β-catenin in an m6A-dependent manner, promoting the proliferation and
metastasis of liver cancer cells8. On the other hand, overexpression of METTL14 in cells can inhibit the EGFR/PI3K/AKT signaling pathway in an m6A methylation-dependent manner, thereby
suppressing tumor metastasis17. METTL14 also regulates the glycolytic pathway and inhibits tumor proliferation through the USP48-SIRT6 pathway7. These findings are consistent with our
bioinformatics analysis results (Fig. 1). When we performed survival analysis using METTL3 and METTL14 as a gene set, we found that METTL3 and METTL14 contributed completely opposite
prognostic risk values in liver cancer (Fig. S2C). Although METTL3 and METTL14 have different prognostic values as components of the m6A complex, overall, they are still associated with poor
prognosis in tumors. To investigate the underlying molecular mechanisms, we also conducted preliminary studies using RNA-seq. We found that METTL3, independent of METTL14, regulates
pathways such as Cell Cycle and DNA damage response (Fig. 3B). The regulatory mechanism of METTL3 independent of METTL14 has been rarely reported. Some studies suggest that cytoplasmic
localized METTL3 can directly bind to the eukaryotic translation initiation factor eIF3h, independent of METTL14. The amino acid residue 155A of METTL3 is a key site for its direct
interaction with eIF3h, forming dense polyribosomes. Ribosome-localized METTL3 enhances mRNA translation through a gap-dependent mechanism and plays an important role in the progression of
lung cancer18. A similar mechanism has been observed in chronic myeloid leukemia19. However, in our study, both at the cellular level and in tumor tissues, METTL3 did not exhibit cytoplasmic
localization (Fig. 4A,C), suggesting that the METTL3-independent function from METTL14 occurs within the nucleus, at least under our experimental conditions. Within the nucleus, the
association of METTL3–METTL14 with the transcriptional complex is an important mechanism for efficient mRNA modification by the “writer” complex. In normal cells, METTL3 and METTL14 form a
complex that faithfully associates with the transcriptional complex. METTL14 binds to trimethylated histone H3K36me313, allowing the coupling of METTL3-METTL14 with the transcriptional
complex, along with specific transcription factors, to link transcription with m6A modification on RNA20. Their binding sites on DNA are highly consistent. However, during cellular
senescence, there is only 13% overlap in the binding of METTL3 and METTL14 on DNA sequences10,11. This suggests that, under specific conditions, the binding of METTL3 and METTL14 to DNA is
not faithful. In acute myeloid leukemia, METTL3 and METTL14 exhibit binding to transcription start sites, but the binding sites of METTL3 and METTL14 are almost entirely distinct21,22.
Researchers speculate that the differential regulatory patterns of METTL3 and METTL14 may be due to differences in the pathways by which the “writer” complex binds to mRNA. However, in our
study, with EC109 esophageal squamous cell carcinoma cells, we found significant differences in the binding partners of METTL3 and METTL14 (Fig. 4). Among the shared binding partners of
METTL3 and METTL14, we identified histone and transcription-related proteins (such as TAF15), which confirms the association between the m6A modification process and transcription. However,
we also found that METTL3 binds to many partners independent of METTL14. This suggests that the large differences in DNA binding sites observed in senescent or cancer cells may be due to
partial dissociation of the METTL3-METTL14 complex or redundancy in the protein abundance of METTL3 compared to METTL14. We also found that METTL3 independently binds to proteins associated
with mRNA splicing, such as U2AF1, HNRNPM, PCBP1, suggesting that METTL3 may regulate cellular processes through binding to the mRNA splicing complex. m6A reader proteins highly associated
with m6A modifications, such as HNRNPA2B123, HNRNPC24, YTHDC125,26, are known to play a widespread role in regulating m6A-dependent alternative splicing. In nascent RNA, 10% of m6A
modification sites are located within introns or exons near the 5ʹ splice site. Rapid depletion of METTL3 eliminates existing m6A modification sites within introns and alters the original
splicing pattern27. However, in our study, the splicing-associated proteins bound by METTL3, such as U2AF128, HNRNPF29, PCBP230, all have binding capabilities at the 3ʹ end. The role of
METTL3 in splicing proteins may not depend on its m6A-writing ability, similar to its binding to eIF3h18. It is worth noting that proteins like HNRNPF and PCBP2 have binding capabilities for
G-rich or C-rich sequences. Further research is needed to investigate the binding and function of METTL3 with splicing proteins. In conclusion, through bioinformatic, transcriptomic, and
proteomic approaches, we reported the differences in expression, prognostic value, regulatory mechanisms, and distribution of METTL3 and METTL14 in tumors. However, there are still many
questions that require further investigation. For example, how much redundancy exists between METTL3 and METTL14? Does the METTL3 functionality independent of METTL14 depend on its
m6A-writing ability? What is the mechanism by which METTL3, independent of METTL14, binds to different complexes? Does it rely on RNA or recruitment by other proteins? These aspects need to
be further investigated. DATA AVAILABILITY The raw data of mRNA-seq has been uploaded to the GEO database (GSE254232), and the proteomic data has been uploaded to the PRIDE database
(PXD048886). REFERENCES * Deng, L. J. _et al._ m6A modification: Recent advances, anticancer targeted drug discovery and beyond. _Mol. Cancer_ 21(1), 52.
https://doi.org/10.1186/s12943-022-01510-2 (2022). Article CAS PubMed PubMed Central Google Scholar * Fang, Z. _et al._ Role of m6A writers, erasers and readers in cancer. _Exp.
Hematol. Oncol._ 11(1), 45. https://doi.org/10.1186/s40164-022-00298-7 (2022). Article CAS PubMed PubMed Central Google Scholar * Nombela, P., Miguel-Lopez, B. & Blanco, S. The role
of m(6)A, m(5)C and Psi RNA modifications in cancer: Novel therapeutic opportunities. _Mol. Cancer_ 20(1), 18. https://doi.org/10.1186/s12943-020-01263-w (2021). Article CAS PubMed
PubMed Central Google Scholar * An, Y. & Duan, H. The role of m6A RNA methylation in cancer metabolism. _Mol. Cancer_ 21(1), 14. https://doi.org/10.1186/s12943-022-01500-4 (2022).
Article CAS PubMed PubMed Central Google Scholar * Vu, L. P. _et al._ The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and
leukemia cells. _Nat. Med._ 23(11), 1369–1376. https://doi.org/10.1038/nm.4416 (2017). Article CAS PubMed PubMed Central Google Scholar * Wu, L., Wu, D., Ning, J., Liu, W. & Zhang,
D. Changes of N6-methyladenosine modulators promote breast cancer progression. _BMC Cancer_ 19(1), 326. https://doi.org/10.1186/s12885-019-5538-z (2019). Article PubMed PubMed Central
Google Scholar * Guan, Q. _et al._ Functions, mechanisms, and therapeutic implications of METTL14 in human cancer. _J. Hematol. Oncol._ 15(1), 13. https://doi.org/10.1186/s13045-022-01231-5
(2022). Article CAS PubMed PubMed Central Google Scholar * Zeng, C., Huang, W., Li, Y. & Weng, H. Roles of METTL3 in cancer: Mechanisms and therapeutic targeting. _J. Hematol.
Oncol._ 13(1), 117. https://doi.org/10.1186/s13045-020-00951-w (2020). Article CAS PubMed PubMed Central Google Scholar * Ma, Z., Li, Q., Liu, P., Dong, W. & Zuo, Y. METTL3
regulates m6A in endometrioid epithelial ovarian cancer independently of METTl14 and WTAP. _Cell Biol. Int._ 44(12), 2524–2531. https://doi.org/10.1002/cbin.11459 (2020). Article CAS
PubMed Google Scholar * Liu, P. _et al._ m(6)A-independent genome-wide METTL3 and METTL14 redistribution drives the senescence-associated secretory phenotype. _Nat. Cell Biol._ 23(4),
355–365. https://doi.org/10.1038/s41556-021-00656-3 (2021). Article CAS PubMed PubMed Central Google Scholar * Liu, Z. _et al._ A methyltransferase-like 14/miR-99a-5p/tribble 2 positive
feedback circuit promotes cancer stem cell persistence and radioresistance via histone deacetylase 2-mediated epigenetic modulation in esophageal squamous cell carcinoma. _Clin. Transl.
Med._ 11(9), e545. https://doi.org/10.1002/ctm2.545 (2021). Article CAS PubMed PubMed Central Google Scholar * Zhou, Y. _et al._ Metascape provides a biologist-oriented resource for the
analysis of systems-level datasets. _Nat. Commun._ 10(1), 1523. https://doi.org/10.1038/s41467-019-09234-6 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Huang, H. _et
al._ Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. _Nature_ 567(7748), 414–419. https://doi.org/10.1038/s41586-019-1016-7 (2019). Article ADS
CAS PubMed PubMed Central Google Scholar * Wang, Y. _et al._ N6-Methyladenosine modification destabilizes developmental regulators in embryonic stem cells. _Nat. Cell Biol._ 16(2),
191–198. https://doi.org/10.1038/ncb2902 (2014). Article CAS PubMed PubMed Central Google Scholar * Zaccara, S., Ries, R. J. & Jaffrey, S. R. Reading, writing and erasing mRNA
methylation. _Nat. Rev. Mol. Cell Biol._ 20(10), 608–624. https://doi.org/10.1038/s41580-019-0168-5 (2019). Article CAS PubMed Google Scholar * Sang, L. _et al._ The m(6)A RNA
methyltransferase METTL3/METTL14 promotes leukemogenesis through the mdm2/p53 pathway in acute myeloid leukemia. _J. Cancer_ 13(3), 1019–1030. https://doi.org/10.7150/jca.60381 (2022).
Article CAS PubMed PubMed Central Google Scholar * Shi, Y., Zhuang, Y., Zhang, J., Chen, M. & Wu, S. METTL14 inhibits hepatocellular carcinoma metastasis through regulating
EGFR/PI3K/AKT signaling pathway in an m6A-dependent manner. _Cancer Manag. Res._ 12, 13173–13184. https://doi.org/10.2147/CMAR.S286275 (2020). Article CAS PubMed PubMed Central Google
Scholar * Choe, J. _et al._ mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. _Nature_ 561(7724), 556–560. https://doi.org/10.1038/s41586-018-0538-8
(2018). Article ADS CAS PubMed PubMed Central Google Scholar * Ianniello, Z. _et al._ New insight into the catalytic -dependent and -independent roles of METTL3 in sustaining aberrant
translation in chronic myeloid leukemia. _Cell Death Dis._ 12(10), 870. https://doi.org/10.1038/s41419-021-04169-7 (2021). Article CAS PubMed PubMed Central Google Scholar * Xu, W. _et
al._ Dynamic control of chromatin-associated m(6)A methylation regulates nascent RNA synthesis. _Mol. Cell_ 82(6), 1156–1168. https://doi.org/10.1016/j.molcel.2022.02.006 (2022). Article
CAS PubMed PubMed Central Google Scholar * Barbieri, I. _et al._ Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. _Nature_ 552(7683), 126–131.
https://doi.org/10.1038/nature24678 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Bertero, A. _et al._ The SMAD2/3 interactome reveals that TGFbeta controls m(6)A mRNA
methylation in pluripotency. _Nature_ 555(7695), 256–259. https://doi.org/10.1038/nature25784 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Alarcon, C. R. _et al._
HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. _Cell_ 162(6), 1299–1308. https://doi.org/10.1016/j.cell.2015.08.011 (2015). Article CAS PubMed PubMed Central
Google Scholar * Liu, N. _et al._ N(6)-Methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. _Nature_ 518(7540), 560–564. https://doi.org/10.1038/nature14234
(2015). Article ADS CAS PubMed PubMed Central Google Scholar * Kasowitz, S. D. _et al._ Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte
development. _PLoS Genet._ 14(5), e1007412. https://doi.org/10.1371/journal.pgen.1007412 (2018). Article CAS PubMed PubMed Central Google Scholar * Roundtree, I. A. _et al._ YTHDC1
mediates nuclear export of N(6)-methyladenosine methylated mRNAs. _Elife_ 6, 311. https://doi.org/10.7554/eLife.31311 (2017). Article Google Scholar * Wei, G. _et al._ Acute depletion of
METTL3 implicates N (6)-methyladenosine in alternative intron/exon inclusion in the nascent transcriptome. _Genome Res._ 31(8), 1395–1408. https://doi.org/10.1101/gr.271635.120 (2021).
Article PubMed PubMed Central Google Scholar * Biancon, G. _et al._ Precision analysis of mutant U2AF1 activity reveals deployment of stress granules in myeloid malignancies. _Mol. Cell_
82(6), 1107–1122. https://doi.org/10.1016/j.molcel.2022.02.025 (2022). Article CAS PubMed PubMed Central Google Scholar * Dominguez, C., Fisette, J. F., Chabot, B. & Allain, F. H.
Structural basis of G-tract recognition and encaging by hnRNP F quasi-RRMs. _Nat. Struct. Mol. Biol._ 17(7), 853–861. https://doi.org/10.1038/nsmb.1814 (2010). Article CAS PubMed Google
Scholar * Chen, X. _et al._ PCBP2 reduced oxidative stress-induced apoptosis in glioma through cGAS/STING pathway by METTL3-mediated m6A modification. _Oxid. Med. Cell Longev._ 2022,
9049571. https://doi.org/10.1155/2022/9049571 (2022). Article CAS PubMed PubMed Central Google Scholar Download references FUNDING This work were supported in part by grants from the
National natural science foundation of China (Grant No. 32000665). Technology Commission Foundation of Shanxi Province (Grant Nos. 20210302124292, 20210302124296, 202203021222315,
20210302124091), Changzhi Medical College doctoral research fund (Grant No. BS202101). Changzhi Medical College Academic Leader Project (Grant No. XS202101). AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Center of Healthy Aging, Changzhi Medical College, Changzhi, 047500, China Bin Du, Pu Wang, Kai Qin, Jinping Zheng & Jia Wang * Central Laboratory of Clinical Research,
Heping Hospital Affiliated to Changzhi Medical College, Changzhi, 047500, China Lingyu Wei * Department of Physiology, Changzhi Medical College, Changzhi, 047500, China Zhen Pei Authors *
Bin Du View author publications You can also search for this author inPubMed Google Scholar * Pu Wang View author publications You can also search for this author inPubMed Google Scholar *
Lingyu Wei View author publications You can also search for this author inPubMed Google Scholar * Kai Qin View author publications You can also search for this author inPubMed Google Scholar
* Zhen Pei View author publications You can also search for this author inPubMed Google Scholar * Jinping Zheng View author publications You can also search for this author inPubMed Google
Scholar * Jia Wang View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Pu Wang is responsible for the overall content as guarantor. Pu Wang
conceived and designed the project. During the revise stage, all work was completed by Bin Du. Kai Qin performed experiments and analyzed the ESCC data. Zhen Pei, Jia Wang, Lingyu Wei and
Jinping Zheng performed experiments, interpreted the data and prepared the figures. Lingyu Wei contributed biopsy samples and pathology analysis. Pu Wang wrote the manuscript. All authors
read and approved the manuscript. CORRESPONDING AUTHOR Correspondence to Jia Wang. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION
PUBLISHER'S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
INFORMATION. SUPPLEMENTARY FIGURE S1. SUPPLEMENTARY FIGURE S2. SUPPLEMENTARY FIGURE S3. SUPPLEMENTARY FIGURE S4. SUPPLEMENTARY TABLE S1. RIGHTS AND PERMISSIONS OPEN ACCESS This article is
licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in
this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's
Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Du, B., Wang, P., Wei, L. _et al._ Unraveling
the independent role of METTL3 in m6A modification and tumor progression in esophageal squamous cell carcinoma. _Sci Rep_ 14, 15398 (2024). https://doi.org/10.1038/s41598-024-64517-3
Download citation * Received: 17 January 2024 * Accepted: 10 June 2024 * Published: 04 July 2024 * DOI: https://doi.org/10.1038/s41598-024-64517-3 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative KEYWORDS * METTL3 * METTL14 * Independent * Proliferation * Splicing