Angiotensin ii participates in mitochondrial thermogenic functions via the activation of glycolysis in chemically induced human brown adipocytes
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Brown adipocytes are potential therapeutic targets for the prevention of obesity-associated metabolic diseases because they consume circulating glucose and fatty acids for heat
production. Angiotensin II (Ang II) peptide is involved in the pathogenesis of obesity- and cold-induced hypertension; however, the mechanism underlying the direct effects of Ang II on human
brown adipocytes remains unclear. Our transcriptome analysis of chemical compound-induced brown adipocytes (ciBAs) showed that the Ang II type 1 receptor (AGTR1), but not AGTR2 and MAS1
receptors, was expressed. The Ang II/AGTR1 axis downregulated the expression of mitochondrial uncoupling protein 1 (_UCP1_). The simultaneous treatment with β-adrenergic receptor agonists
and Ang II attenuated _UCP1_ expression, triglyceride lipolysis, and cAMP levels, although cAMP response element-binding protein (CREB) phosphorylation was enhanced by Ang II mainly through
the protein kinase C pathway. Despite reduced lipolysis, both coupled and uncoupled mitochondrial respiration was enhanced in Ang II-treated ciBAs. Instead, glycolysis and glucose uptake
were robustly activated upon treatment with Ang II without a comprehensive transcriptional change in glucose metabolic genes. Elevated mitochondrial energy status induced by Ang II was
likely associated with _UCP1_ repression. Our findings suggest that the Ang II/AGTR1 axis participates in mitochondrial thermogenic functions via glycolysis. SIMILAR CONTENT BEING VIEWED BY
OTHERS TREATMENT WITH ATRIAL NATRIURETIC PEPTIDE INDUCES ADIPOSE TISSUE BROWNING AND EXERTS THERMOGENIC ACTIONS IN VIVO Article Open access 31 August 2021 CAPSAICIN DIRECTLY PROMOTES
ADIPOCYTE BROWNING IN THE CHEMICAL COMPOUND-INDUCED BROWN ADIPOCYTES CONVERTED FROM HUMAN DERMAL FIBROBLASTS Article Open access 22 April 2022 LACK OF TRPV1 AGGRAVATES OBESITY-ASSOCIATED
HYPERTENSION THROUGH THE DISTURBANCE OF MITOCHONDRIAL CA2+ HOMEOSTASIS IN BROWN ADIPOSE TISSUE Article Open access 18 January 2022 INTRODUCTION Excess energy intake has increased the
incidence of obesity and type 2 diabetes epidemics. Obesity is a serious risk factor for pathogenic metabolic alterations, such as impaired glucose tolerance, insulin resistance, and chronic
inflammation1. Brown adipose tissue (BAT) is responsible for adaptive non-shivering thermogenesis in mammals2. Brown adipocytes specialise in heat generation by consuming blood glucose and
free fatty acids for cold acclimation3. Cold circumstances activate brown adipocytes by the secretion of norepinephrine (NE), an endogenous β-adrenergic receptor (βAR) agonist, from the
terminals of sympathetic nerves3. Heat production is conferred by mitochondrial uncoupling protein 1 (UCP1) located on the inner membrane4. UCP1 relieves the electron proton gradient for
heat generation, which is uncoupled from adenosine triphosphate (ATP) synthesis. BAT contributes not only to adaptive thermogenesis but also to the improvement of symptoms of metabolic
syndrome by fuelling circulating blood glucose, free fatty acids, branched-chain amino acids, and acylcarnitin5,6. BAT functions as a metabolic sink and helps improve blood glucose,
triglycerides, and high-density lipoprotein levels, thereby reducing the risk of cardiometabolic diseases7. The renin-angiotensin system (RAS) regulates blood pressure, fluid, and
electrolyte homeostasis and is involved in many physiological events in diverse organs8. The adipose tissue is known to serve as the primary source of angiotensinogen (AGT)9. Obesity is
correlated with increased plasma levels of AGT, renin, angiotensin-converting enzyme (ACE), and aldosterone10. Human adipose tissue expresses the RAS components required for the
responsiveness to angiotensin II (Ang II)11. Locally derived or circulating Ang II negatively regulates adipocyte differentiation and functions12,13. The anti-adipogenic effects of Ang II
are due to the activation of the extracellular signal-regulated kinase (ERK) in the mitogen-activated protein kinase (MAPK) signalling pathway through the type 1 receptor, AGTR114,15. In
contrast, AGTR1-knockout rats showed ameliorated diet-induced obesity, elevated adipose lipolysis, and fatty acid oxidation16. This was caused by the activation of the cAMP/protein kinase A
(PKA) pathway, which overlaps with the thermogenesis pathway initiated by βAR receptors17. In obesity, the RAS hyperactivation of the classical arm mediated by Ang II/AGTR1 is exacerbated
and associated with obesity-induced hypertension, insulin resistance, and inflammation18,19. In humans, plasma AGT and leptin levels, blood pressure, and insulin secretion in response to
oral glucose load are positively correlated with body mass index, suggesting a close relationship between the RAS and obesity20. Clinical epidemiological studies have shown that chronic or
intermittent cold exposure is a risk factor for hypertension and cardiovascular diseases21,22. Cold temperatures activate the sympathetic nervous system (SNS), leading to cold-induced
hypertension (CIH) via the RAS23,24. The elevation of blood pressure during CIH is beneficial for circulatory function and metabolic rate during non-shivering thermogenesis25,26,27.
Cold-induced increases in systolic blood pressure are largely abolished by treatment with losartan, a potent AGTR1 inhibitor28. An increase in hypothalamic AGTR1 expression is involved in
the development of CIH, whereas the genetic ablation of AGTR1 attenuates CIH29. Notably, cold exposure elevated blood Ang II and NE levels in rats and human subjects30,31,32, implying that
Ang II may be involved in the regulation of cold-induced thermogenesis. Ang II treatment increased Ang II content in rat interscapular BAT and facilitated the presynaptic release of NE from
cold-exposed rat BAT33. Several reports have indicated that Ang II facilitated the synthesis and uptake of NE in adipose tissue through AGTR134,35, indicating an association between Ang II
and thermogenesis. Gene expression analysis indicated that thermogenic and energy metabolic genes, including _UCP1_, increased in the adipose tissues of chronic cold-exposed mice, which
might be involved in the development of CIH36. Thus, Ang II is closely associated with cold-induced thermogenesis; however, the molecular mechanism underlying the direct action of Ang II in
brown adipocytes has not been fully elucidated. To overcome the difficulty of leveraging a sufficient amount of pure human brown adipocytes, we developed a method for the chemical conversion
of the primary cultures of human dermal fibroblasts (HDFs) into brown adipocytes37. Chemical compound-induced brown adipocytes (ciBAs) were converted using a cocktail (RoFB) consisting of
rosiglitazone, forskolin, and bone morphogenetic protein 7 (BMP7) in serum-free conditions, as previously reported38. Our previous study indicated that ciBAs expressed UCP1 more robustly
than adipocytes differentiated from mesenchymal stem cells (MSCs)39. In addition, our transcriptome analysis indicated that a specific set of the RAS components was expressed in human brown
adipocyte models, including ciBAs39,40. Our results suggest that the Ang II/AGTR1 axis contributes to thermogenic functions by mediating cellular lipid and glucose metabolism in human brown
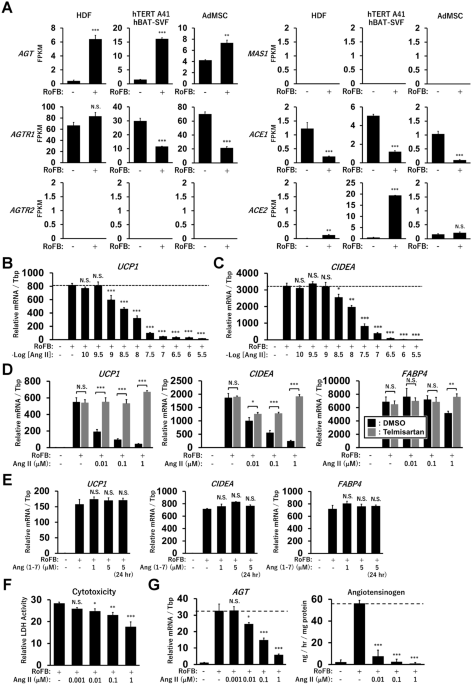
adipocytes. RESULTS ANG II TREATMENT REPRESSES UCP1 EXPRESSION IN HUMAN BROWN ADIPOCYTE MODELS Our previous transcriptome analysis showed that a specific set of the RAS components, including
_AGT_, _AGTR1_, and _ACE1_, was expressed in human brown adipocyte models derived from HDFs, immortalised human preadipocytes (hTERT A41hBAT-SVF), and adipose tissue-derived MSCs (AdMSCs)
(Fig. 1A). In contrast, _AGTR2_ and the Mas receptor (_MAS1_) exhibited low expression levels, suggesting that the classical ACE1/Ang II/AGTR1 axis, but not the counter-regulatory ACE2/Ang
(1-7)/MAS1 axis, could be a major RAS pathway in these adipocytes, including ciBAs. Although renin (_REN_) expression was limited to these adipocytes, the (pro)renin receptor, _ATP6AP2_, was
abundantly expressed (Supplementary Fig. S1A). The low expression of chymase (_CAM1_) indicated that the ACE-independent synthesis of Ang II was negligible, whereas neprilysin (_NEP_)
expression implied the possible synthesis of ANG (1–7) peptide from Ang I peptide. Next, to evaluate the effects of Ang II on thermogenic gene expression and brown adipogenesis, cells were
continuously treated with Ang II during the chemical conversion of HDFs into ciBAs. Ang II strongly reduced the expression of _UCP1_ and _CIDEA_, another brown adipocyte-enriched gene, in a
dose-dependent manner at concentrations more than 1 nM (Fig. 1B,C). Incubation with Ang II for more than 3 days was required to elicit repressive effects on _UCP1_ expression in ciBAs
(Supplementary Fig. S1B–D). The repressive effects of Ang II were detected in ciBAs derived from other lines of HDFs and adipocytes derived from hTERT A41hBAT-SVF; however, relatively lower
effects were detected in adipocytes derived from AdMSCs (Supplementary Fig. S2A–C). The repression of _UCP1_, _CIDEA_, and _FABP4_ expression by Ang II was reversed by the addition of
telmisartan, a potent inhibitor of AGTR1 (Fig. 1D). The expression of the adipocyte-enriched gene _FABP4_ was slightly repressed at high concentrations of Ang II. The counter-regulatory
peptide, Ang (1–7), an endogenous agonist of MAS1, had little effect on the expression, consistent with the low expression level of MAS1 (Fig. 1E). In addition, treatment with MLN-4760, a
potent ACE2 inhibitor, had little effect on the repressive effects of Ang II (Supplementary Fig. S2D). Cytotoxicity was reduced in ciBAs treated with Ang II in a dose-dependent manner (Fig.
1F). Ang II treatment suppressed the expression and secretion of AGT in a dose-dependent manner (Fig. 1G). These results indicated that the Ang II treatment robustly repressed thermogenic
_UCP1_ transcription through the Ang II/AGTR1 axis in human brown adipocyte models. ANG II TREATMENT REDUCES TRIGLYCERIDE LIPOLYSIS Lipid metabolism was characterised in Ang II-treated
ciBAs. Similar to _UCP1_ mRNA levels, protein expression was reduced by Ang II treatment in a dose-dependent manner (Fig. 2A). The protein levels of ATGL, a rate-limiting enzyme in adipocyte
lipolysis, and CEBPA, a critical regulator of adipocyte differentiation, were repressed by Ang II treatment. Immunocytochemical analysis showed that fluorescent signals for lipid droplets
and UCP1 protein were reduced by Ang II (Fig. 2B,C and Supplementary Fig. S3A). Consistently, cellular triglyceride storage was decreased by Ang II treatment (Fig. 2D). Notably, glycerol
secretion in Ang II-treated ciBAs was robustly reduced, indicating that lipolysis was suppressed (Fig. 2E). The reduced glycerol secretion was reversed by simultaneous treatment with
telmisartan. In addition, cellular glycerol levels were reduced by Ang II, implying that the reduction of glycerol secretion was unlikely to be due to increased glycerol uptake for recycling
(Fig. 2F). The reduced phosphorylation of hormone-sensitive lipase (HSL) supported the reduced lipolysis in Ang II-treated ciBAs (Fig. 2G). The reduced phosphorylation of HSL was partially
reversed by telmisartan treatment (Fig. 2H). Next, to address how Ang II regulates lipid metabolism, related signalling pathways were analysed. Ang II treatment reduced the phosphorylation
of cAMP responsive element-binding protein (CREB), which was reversed by telmisartan treatment (Fig. 2I). In addition, Ang II enhanced the phosphorylation of the p38 and ERK MAPK signalling
pathways. These results indicated that continuous treatment with Ang II reduced lipolysis through changes in several signalling pathways in ciBAs. TRANSIENT TREATMENT WITH ANG II INHIBITS
ΒAR-MEDIATED UCP1 INDUCTION AND LIPOLYSIS Under cold conditions, blood Ang II and NE levels increase via the activation of the SNS30,31,32. UCP1 expression and lipid lipolysis are activated
for thermogenesis through the cAMP signalling pathway initiated by the activation of βAR by NE41. To evaluate the effects of Ang II on βAR-mediated thermogenesis, Ang II and either
isoproterenol (Iso) or NE were simultaneously administered to ciBAs (Fig. 3A,B). The induced _UCP1_ expression was partially inhibited by treatment with Ang II, and additional treatment with
telmisartan reversed this inhibition. Iso-induced glycerol secretion and cellular cAMP levels were also reduced by Ang II treatment, which was reversed by the additional treatment with
telmisartan (Fig. 3C,D). In contrast, transient treatment with Ang II further enhanced Iso-induced CREB phosphorylation (Fig. 3E). Transient treatment with Ang II increased p38 and ERK
phosphorylation. Although continuous treatment reduced CREB phosphorylation, transient treatment was confirmed to robustly enhance the phosphorylation (Fig. 3F). In contrast, HSL
phosphorylation was reduced by transient treatment (Fig. 3G). To identify upstream signalling pathways responsible for CREB phosphorylation by Ang II, several kinase inhibitors were
pre-incubated before Ang II treatment. PD98059, an inhibitor of the ERK MAPK pathway, caused a slight reduction in the phosphorylation (Fig. 3H). Pre-incubation with SB202190, an inhibitor
of the p38 MAPK pathway, also slightly decreased this effect, whereas Go6983, a potent inhibitor of pan-protein kinase C (PKC), further reduced it (Fig. 3I). KN-62, an inhibitor of
calmodulin-dependent protein kinase II (CaMKII), had little effect on the phosphorylation. In addition, H89, a potent inhibitor of PKA, did not strongly affect the Ang II-mediated CREB
phosphorylation, whereas H89 repressed Iso-mediated CREB phosphorylation (Fig. 3J). These results indicated that Ang II treatment inhibited βAR-mediated UCP1 expression and lipolysis by
partially interfering with the thermogenesis pathway through AGTR1. ANG II TREATMENT ENHANCES MITOCHONDRIAL OXYGEN CONSUMPTION RATE (OCR) THROUGH THE ACTIVATION OF GLYCOLYSIS Despite reduced
lipolysis by Ang II, OCR was increased by continuous treatment with Ang II (Fig. 4A,B). The OCR increased from 0.001 μM and almost reached a plateau at concentrations greater than 0.01 μM.
The OCR, corresponding to basal and maximal respiration, ATP production, and proton leak was elevated by Ang II treatment (Fig. 4C). The percent ratio of proton leak in basal respiration was
only enhanced in ciBAs treated with Ang II at 1 μM, indicating that uncoupling efficiency was largely consistent in Ang II-treated ciBAs (Fig. 4D). The percent ratio of ATP production in
maximum respiration was slightly reduced, indicating that the portion utilised by maximum respiratory capacity was almost unchanged by Ang II treatment. Notably, extracellular acidification
rate (ECAR) strongly increased under both basal and oligomycin-treated conditions (Fig. 4E). Ang II-treated ciBAs were metabolically active and showed increase in both OCR and ECAR (Fig.
4F). ECAR immediately increased upon the addition of Ang II during measurement, while simultaneous treatment with telmisartan largely repressed this increase (Fig. 4G). Importantly, the
addition of Ang II enhanced the OCR corresponding to both basal respiration and proton leak, compared with that in the untreated ciBAs (Fig. 4H). Mitochondrial staining with two different
types of fluorescent probes indicated that Ang II enhanced the mitochondrial content in a dose-dependent manner (Fig. 4I and Supplementary Fig. S3B). The increased ratio of mitochondrial DNA
to nuclear DNA indicated increased mitochondria in Ang II-treated ciBAs (Fig. 4J). Consistent with enhanced OCR, mitochondrial membrane potential (MMP) was elevated in Ang II-treated ciBAs
(Fig. 4K and Supplementary Fig. S3C). These results indicated that Ang II robustly and rapidly promoted glucose utilisation through AGTR1, which contributes to coupled and uncoupled
mitochondrial respiration in ciBAs. To further characterise glucose metabolic processes activated by Ang II, a glycolysis stress assay was performed using glucose, oligomycin, and
2-deoxy-D-glucose (2-DG) in ciBAs cultured under glucose-free conditions. The addition of glucose increased glycolysis, while oligomycin maximised this rate by inhibiting mitochondrial ATP
production. The continuous treatment with Ang II at 0.01 and 0.1 μM strongly enhanced ECAR compared to that in the untreated ciBAs (Fig. 5A). The ECAR, corresponding to glycolysis and
glycolytic capacity, was elevated by Ang II treatment (Fig. 5B). The addition of Ang II to the control ciBAs during measurement immediately increased ECAR (Fig. 5C), which was inhibited by
simultaneous treatment with telmisartan (Fig. 5D). Consistent with this observation, Ang II and telmisartan increased and inhibited glycolysis and glycolytic capacity, respectively (Fig.
5E). In addition, the OCR was enhanced upon treatment with Ang II, which was reversed by telmisartan treatment (Supplementary Fig. S4A,B). Glucose uptake, evaluated by a fluorescent glucose
probe, was robustly increased by both transient and continuous treatment with Ang II (Fig. 5F). The phosphorylation of the serine/threonine kinase (AKT) in the insulin signalling pathway was
reduced by both continuous and transient treatments with Ang II, which was reversed by telmisartan (Fig. 5G and Supplementary Fig. S5). These results support the conclusion that Ang II
treatment coordinates cellular and mitochondrial energy metabolism by activating glycolysis and glucose uptake in ciBAs. ANG II TREATMENT DOES NOT ACTIVATE THE TRANSCRIPTION OF GLUCOSE
METABOLIC GENES To address how Ang II treatment alters metabolic gene expression, a genome-wide transcriptome analysis was performed. A comparison of the RNA-Seq data between control (RoFB)
and AngII-treated ciBAs (RoFB + Ang II) revealed 815 upregulated and 985 downregulated differentially expressed genes (DEGs) (Fig. 6A). The smear and volcano plots showed that DEGs with over
two-fold changes (FCs) were appropriately distributed with widespread counts per million (CPM) and _p_-values (Supplementary Fig. S6A). Venn diagrams show that 787 upregulated and 575
downregulated DEGs overlapped (Supplementary Fig. S6B). Multidimensional scaling analysis visually suggested that Ang II treatment affected the transcriptome of ciBAs (Fig. 6B). However,
gene ontology (GO) enrichment analysis showed that the upregulated DEGs were not directly associated with glucose or lipid metabolism (Fig. 6C). Ang II treatment downregulated a series of
muscle-related genes. Heat maps represented that Ang II treatment did not consistently increase the expression of glucose metabolism-related genes (Fig. 6D), whereas many muscle-related
genes were repressed (Fig. 6E and Supplementary Table S1). Although several metabolic genes involved in the tricarboxylic acid (TCA) cycle and fatty acid β-oxidation were rather reduced
(Supplementary Fig. S6C), the expression of genes involved in fatty acid synthesis was slightly increased by Ang II treatment (Fig. 6F). Quantitative real-time polymerase chain reaction
(qRT-PCR) analysis confirmed that the expression of glucose metabolic genes, such as _HK1_, _HK2_, _PFKFB3_, _PDHA1_, _LDHB_, _G6PD_, _SLC2A1_, _SLC2A4_, _PDHA1_, _PC_, and _CS_, was not
robustly increased by both transient and continuous treatment with Ang II (Fig. 6G). These results indicated that the activation of glycolysis by Ang II was unlikely to be due to
transcriptional changes in metabolic genes. DISCUSSION In this study, we revealed that Ang II treatment affected thermogenic functions and cellular energy metabolism in ciBAs (Fig. 7). Ang
II treatment repressed the transcription of _UCP1_ and _CIDEA_ as well as triglyceride lipolysis and storage. Reduced lipolysis by Ang II was associated with the downregulation of ATGL,
CEBPA, and HSL phosphorylation. Despite this, Ang II-treated ciBAs showed increased mitochondrial OCR and MMP along with elevated mitochondrial contents. Notably, Ang II treatment rapidly
and highly activated glycolysis and glucose uptake, which likely promoted metabolic reprogramming from lipolysis to glycolysis in ciBAs. Importantly, the activation of glycolysis was
associated with an increase in coupled and uncoupled mitochondrial respiration. The effects of Ang II on glucose and lipid metabolism were exerted through the Ang II/AGTR1 axis, and
treatment with telmisartan largely abolished these effects. We previously proposed a feedback regulation between mitochondrial energy status and thermogenic _UCP1_ transcription in
ciBAs40,41. MMP induced by enriched free fatty acids, mimicking obese conditions, downregulated _UCP1_ transcription likely to avoid excess heat generation. In contrast, the depletion of
free fatty acids induced _UCP1_ expression likely to compensate for uncoupled proton leak under low MMP conditions. The feedback regulation is considered beneficial for coordinating the
thermogenic proton leak activity mediated by UCP1 in brown adipocytes. Our study also found that the Ang II-activated mitochondrial energy status led to the repression of _UCP1_
transcription, similar to the feedback regulation described above. These results may partially explain the obesity-associated repression of _UCP1_ expression under the hyperactivation of the
RAS during obesity19,41. Our previous transcriptome analysis revealed that a specific set of the RAS components, including AGT and AGTR1, was expressed in multiple human adipocyte models
derived from HDFs, hTERT A41hBAT-SVF, and AdMSCs (Fig. 1). Treatment with telmisartan under exogenous Ang II-free conditions did not influence the expression of _UCP1_ and _CIDEA_, implying
that Ang II was not locally produced from AGT in ciBAs. Ang II is deactivated by ACE2 and converted into angiotensin 1-7 peptide, Ang (1-7), acting as an agonist of MAS1. The
counter-regulatory ACE2/Ang (1-7)/MAS1 axis is known to have beneficial effects on diet-induced weight gain, adipocyte browning, thermogenesis, and metabolic complications via the activation
of AKT and inhibition of the ERK MAPK signalling pathway14,42. Mice with whole-body knockout of either ACE2 or MAS1 displayed metabolic abnormalities, whereas either overexpression of ACE2
or continuous infusion of Ang (1-7) peptide improved blood glucose, lipid, and energy expenditure, along with enhanced adipocyte browning in WAT43. Another recent study indicated that
hamster BAT was activated by the third ventricular injection of Ang (1-7) peptide through the Mas1 receptor in brain regions44. In this study, the treatment with Ang (1-7) had little effect
on the expression levels of _UCP1_, _CIDEA_, and _FABP4_, suggesting that the counter-regulatory axis is not functional in ciBAs. This was consistent with the observation that MAS1
expression was negligible in ciBAs and the other models of human adipocytes. These results indicated that the classical Ang II/AGTR1 axis may be the major RAS pathway in human brown
adipocytes. The short-term treatment with Ang II inhibited βAR-mediated induction of _UCP1_ expression, lipolysis, and cAMP levels (Supplementary Fig. S7A). Previous studies have reported
that the Gαi protein is one of the effectors of AGTR117. Gαi reduces cAMP levels by inhibiting the adenylyl cyclase activity in response to Ang II treatment, suggesting that Ang II effects
partially overlapped with the thermogenesis pathway45. This is further supported by the observation that continuous treatment with Ang II decreased the phosphorylation of HSL and CREB as
downstream events in the cAMP signalling pathway (Supplementary Fig. S7B)41. However, transient treatment with Ang II enhanced the phosphorylation of CREB, but not of HSL. The canonical
signalling of the AGTR1 receptor also couples to the Gαq protein to activate phospholipase C (PLC), resulting in the activation of PKC by the increase of Ca2+ concentrations and
diacylglycerol (DAG) (Supplementary Fig. S7A)46,47. The potent PKC inhibitor, Go6983, robustly reduced CREB phosphorylation by Ang II, suggesting that AGTR1 positively and negatively
contributes to the phosphorylation of CREB through Gαq and Gαi, respectively. In addition, our results indicated that the PKA pathway was not closely associated with Ang II-mediated CREB
phosphorylation. Similar to other reports15,17, this study indicated that the phosphorylation of p38 and ERK in the MAPK signalling pathway was increased by both transient and continuous
treatment with Ang II. The results obtained using each inhibitor indicated that they both partially contributed to Ang II-induced CREB phosphorylation. It has been reported that Ang II is
involved in the pathogenesis of insulin resistance and cardiovascular diseases47,48. Adipose tissue-specific overexpression of AGT or Ang II infusion caused systemic insulin resistance49.
However, Ang II did not alter the binding and activity of the insulin receptor50,51. Mechanistically, Ang II negatively regulates the insulin signalling cascade, including the tyrosine
phosphorylation of the insulin receptor and insulin receptor substrates, phosphatidylinositol 3-kinase (PI3K), and AKT52,53. This study found that Ang II robustly enhanced glycolytic flux
and glucose uptake in ciBAs. AKT phosphorylation was repressed by Ang II through AGTR1, implying that Ang II-activated glycolysis is unlikely to occur through the activation of the insulin
signalling pathway. In previous studies, Ang II inhibited adipocyte lipolysis by approximately 20–25% in human subjects in an AGTR1-dependent manner54,55. This suggests that metabolic
reprogramming from lipolysis to glycolysis is promoted by Ang II treatment. Ang II-activated glycolysis contributes to the elevation of mitochondrial OCR, suggesting that glycolytic pyruvate
may be used for the anaplerosis of the TCA cycle. However, transcriptome analysis of Ang II-treated ciBAs revealed that the transcription of glucose and lipid metabolic genes remained
largely unchanged, despite the activation of glycolysis. Although a series of muscle-related genes were consistently repressed by Ang II, their association with either glycolysis or _UCP1_
expression remains unclear. Rapid response to Ang II appears to be mediated by post-translational and/or post-transcriptional changes that regulate the glycolytic pathway through AGTR1.
Further research is required to clarify the precise molecular mechanisms underlying the activation of glycolysis by Ang II. To date, the regulation of glycolysis and glucose uptake by Ang II
has not been extensively examined in brown adipocytes. Therefore, this study provides additional insights into this mechanism and its association with the thermogenic functions of human
brown adipocytes. The elevated mitochondrial energy status through the Ang II/AGTR1 axis may be beneficial for the efficient consumption of blood glucose for heat production in brown
adipocytes. Although angiotensin-converting enzyme inhibitors and Ang II receptor blockers are expected to prevent type 2 diabetes and obesity-associated metabolic disorders47,48,56, this
study implies that they might affect some aspects of systemic glucose metabolism through brown adipocytes and other metabolic cells. Importantly, cold exposure potentially elevates
circulating Ang II and NE levels along with the SNS activation30,31,32. This study suggests the possible contribution of Ang II to adaptive thermogenesis in human brown adipocytes. We
previously reported that treatment with capsaicin directly activated UCP1 expression along with other browning features in ciBAs57. Thus, the thermogenic functions of ciBAs can be
distinctively regulated by bioactive molecules through cell surface receptors, such as AGTR1 and transient receptor potential vanilloid 1 (TRPV1). Insights obtained from ciBAs are supportive
of uncovering the molecular mechanism of the action targeting human brown adipocytes. METHODS CELL CULTURE Fibroblasts derived from a human subject aged 38 years (HDF38) (PromoCell,
Heidelberg, Germany) were used for this study39. The commercial human cells have been approved for in vitro research use only. All experimental procedures for cell cultures were conducted in
accordance with the general guidelines of the Kyoto Prefectural University of Medicine. Approximately 1.5 × 105 cells were seeded in a 35-mm dish with high-glucose Dulbecco's modified
Eagle medium (DMEM) (11995-065, Gibco, MA, USA) supplemented with 10% fetal bovine serum (FBS) (HyClone, UT, USA) and penicillin/streptomycin (Gibco). After reaching 80–90% confluence, the
medium was changed to start the direct conversion into ciBAs with the serum-free brown adipogenic medium (SFBAM), either with or without the chemical cocktail RoFB, which consisted of 1 μM
rosiglitazone (FUJIFILM Wako), 7.5 μM forskolin (FUJIFILM Wako), and 20 ng/mL human recombinant BMP7 (FUJIFILM Wako) for 3 weeks, as reported previously38. Angiotensin II (015-27911,
FUJIFILM Wako) was added in combination with RoFB during conversion at the indicated concentrations. The medium was changed every 3 days. Angiotensin (1–7) peptide (A9202, Sigma-Aldrich, MO,
USA), telmisartan (205-20691, FUJIFILM Wako), MLN-4760 (31,327, Cayman Chemical, MI, USA), isoproterenol (I0260, Tokyo Chemical Industry, Tokyo, Japan), norepinephrine (A0906, Tokyo
Chemical Industry), PD98059 (FUJIFILM Wako), SB202190 (FUJIFILM Wako), Go6983 (FUJIFILM Wako), and KN-62 (Cayman Chemical) were used to treat ciBAs as indicated. QRT-PCR Total RNA was
extracted from control fibroblasts and ciBAs cultured under each experimental condition using the FastGene RNA Basic Kit (Nippon Genetics, Tokyo, Japan). Reverse transcription was conducted
using ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO, Osaka, Japan). qRT-PCR analysis was performed using Power SYBR Green PCR Master Mix (Applied Biosystems, MA, USA). The
reactions were carried out in triplicate under the following conditions: 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 60 s at 60 °C. All results were normalised to _TBP_ mRNA
levels. Primer sequences used for qRT-PCR are listed in Supplementary Table S2. Unless otherwise indicated, the average of the three biological replicates was calculated. IMMUNOBLOTTING
Total protein was extracted from control fibroblasts, ciBAs, and ciBAs treated with Ang II and/or small molecules using RIPA buffer (FUJIFILM Wako) supplemented with a phosphatase inhibitor
cocktail (FUJIFILM Wako) and protease inhibitor cocktail (FUJIFILM Wako). The extracted proteins were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis using a 10% gel
concentration and transferred to a polyvinylidene fluoride membrane (Thermo Fisher Scientific, MA, USA). The membranes were blocked with 3% skim milk followed by incubation with antibodies
against UCP1 (MAB6158, R&D systems, MN, USA), ATGL (#2138, Cell Signaling Technology, MA, USA), CEBPA (#8178, Cell Signaling Technology), β-Actin (A5316, Sigma-Aldrich), P-HSL (#18381,
Cell Signaling Technology), HSL (#4107, Cell Signaling Technology), P-CREB (#9198, Cell Signaling Technology), CREB (#9197, Cell Signaling Technology), P-p38 (#4511, Cell Signaling
Technology), p38 (#8690, Cell Signaling Technology), P-ERK1/2 (#4370, Cell Signaling Technology), ERK1/2 (#4695, Cell Signaling Technology), P-AKT (#4060, Cell Signaling Technology), and AKT
(#4691, Cell Signaling Technology) overnight at 4 °C or for 2 h at room temperature. Membranes were then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse
secondary antibodies (Cell Signaling Technology) for 1 h at room temperature. Immunoreactive bands were detected using Immobilon Western Chemiluminescent HRP Substrate (Merck Millipore,
Darmstadt, Germany). The intensity of each band was quantified via densitometry using ImageJ software (version 1.52; National Institutes of Health, Bethesda, MD, USA). Experiments were
independently performed at least twice. IMMUNOCYTOCHEMISTRY The cells were preincubated with 1 µM Lipi-Red (Dojindo, Kumamoto, Japan) for 30 min at 37 °C in 5% CO2, according to the
manufacturer’s instructions. Subsequently, the cells were fixed with 4% paraformaldehyde for 10 min. After washing with phosphate-buffered saline (PBS), the cells were incubated in PBS
containing 0.1% Triton X-100 for 5 min. Then, they were blocked with PBS containing 3% skim milk for 1 h at room temperature and incubated again with UCP1 antibody (ab10983, Abcam,
Cambridge, UK) at 1/2000 dilution overnight at 4 °C. After washing with PBS, the cells were incubated with Alexa Fluor 488 donkey anti-rabbit IgG antibody (Invitrogen, CA, USA) for 1 h at
room temperature. Subsequently, cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) solution (Dojindo). All images were obtained using a BZ-X710-All-in-One Fluorescence
Microscope (Keyence, Osaka, Japan) with a 20X objective lens (CFI Plan Fluor 20X, Nikon, Tokyo, Japan). All scale bars represent 200 μm. The areas of Lipi-Red and UCP1 staining were
quantified using ImageJ software from at least five different optical sections. MEASUREMENT OF MITOCHONDRIA CONTENTS AND MEMBRANE POTENTIALS For MMP dependent- and independent-staining,
ciBAs were stained by MitoTracker® Red CM-H2XRos (Thermo Fisher Scientific, DE, USA) and CellLight® Mitochondria-GFP, BacMam 2.0 (C10600, Invitrogen), respectively. In brief, the cells were
pre-incubated with CellLight reagent for 1 d and with MitoTracker for 30 min at 37 °C in a 5% CO2 before the cells were fixed with 4% paraformaldehyde for 10 min. MMPs in control fibroblasts
and ciBAs were evaluated using the MT-1 MitoMP Detection kit (MT13, Dojindo). The cells were treated with the MT-1 dye for 30 min at 37 °C in 5% CO2 incubator, in accordance with the
instructions. Then, the cells were fixed with 4% paraformaldehyde for 10 min. All the images were captured using a BZ-X710-All-in-One Fluorescence Microscope. The area of staining was
quantified from at least five different optical sections using ImageJ software. MEASUREMENT OF GLUCOSE UPTAKE To quantify glucose uptake, each ciBA was treated with a fluorescent glucose
uptake probe (Glucose Uptake Assay Kit-Green, 341-09821, Dojindo) according to the manufacturer’s instructions. Briefly, the cells were washed twice with DMEM without glucose or serum. After
washing, the probe was incubated for 15 min at 37 °C in 5% CO2. After washing twice with an ice-cold solution, the cells were incubated at room temperature for 5 min in the same solution.
After replacing the solution once, images were captured using a BZ-X710-All-in-One Fluorescence Microscope. The staining was quantified as described above. QUANTIFICATION OF MITOCHONDRIAL
DNA Total genomic DNA was extracted from control and Ang II-treated ciBAs using NucleoSpin Tissue (TaKaRa Bio). Mitochondrial DNA (mtDNA) copy numbers were measured by qPCR using the Power
SYBR Green PCR Master Mix. Each mtDNA amount was normalised to the corresponding nuclear DNA (nuDNA) level. Primer sequences used for the quantification of mitochondrial and nuclear DNA were
as follows: mtDNA-Fwd, ACACCCTCCTAGCCTTACTAC; mtDNA-Rev, GATATAGGGTCGAAGCCGC; nuDNA-Fwd, AGGGTATCTGGGCTCTGG; and nuDNA-Rev, GGCTGAAAAGCTCCCGATTAT58. The average of three biological
replicates was calculated. MEASUREMENT OF OCR AND ECAR To measure the mitochondrial OCR, ciBAs were prepared in a 96-well plate under different experimental conditions for 3 weeks. Before
measurement, cells were washed and incubated with non-buffered DMEM supplemented with 25 mM glucose, 2 mM glutamine, and 1 mM pyruvate at 37 °C in a non-CO2 incubator for 1 h. The OCR was
measured using the Seahorse XF96 extracellular flux analyer (Seahorse Bioscience Inc., MA, USA) according to the manufacturer’s instructions. During the analysis, oligomycin, carbonyl
cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), and antimycin A/rotenone were added to each well using an injection apparatus at final concentrations of 2, 0.5, and 0.5 μM, respectively.
The OCR corresponding to each mitochondrial parameter was determined by subtracting the antimycin A/rotenone-insensitive OCR values from other OCR values. To evaluate the rapid effects of
Ang II on the OCR and ECAR, Ang II was added to each well via an injection apparatus during measurement. For measurement of glycolysis flux, the cells were incubated in non-buffered DMEM
without glucose and pyruvate for 1 h at 37 °C in a non-CO2 incubator. ECAR was measured using the Flux Analyzer by adding glucose (Sigma-Aldrich), oligomycin (Sigma-Aldrich), and
2-deoxy-D-glycose (Tokyo Chemical Industry) via an injection apparatus during the measurement at final concentrations of 10 mM, 5 μM, and 50 mM, respectively. The ECAR corresponding to
glycolysis and glycolytic capacity was determined by subtracting the 2-DG-insensitive ECAR values from the other ECAR values before and after oligomycin treatment, respectively. MEASUREMENT
OF GLYCEROL, TRIGLYCERIDES, CAMP, AGT, AND CYTOTOXICITY Cell culture supernatants and extracted cellular lysates were collected from the control fibroblasts, ciBAs, and ciBAs treated with
Ang II, telmisartan, or Iso. The amounts of free glycerol and triglycerides were measured using Free Glycerol Assay Kit (ab65337, Abcam) and Triglyceride Assay kit (Ab65336, Abcam),
respectively, according to the manufacturer’s instructions. cAMP levels in each ciBA sample were measured using a competitive ELISA method (cAMP Assay Kit, ab65355, Abcam). The secretion of
AGT from ciBAs into the supernatant was quantified using a sandwich ELISA (Human Total Angiotensinogen Assay Kit, Immuno-Biological Laboratories, Gunma, Japan). To evaluate the cytotoxicity
of Ang II treatment, the activity of lactate dehydrogenase (LDH) released into the culture supernatants was measured using Cytotoxicity LDH Assay Kit-WST (Dojindo) according to the
manufacturer’s instructions. Glycerol, triglycerides, cAMP, and AGT levels were normalised to the total amounts of protein in each cell culture. All the experiments were performed in
triplicates. Experiments were independently performed at least twice. RNA SEQUENCING (RNA-SEQ) ANALYSIS RNA-Seq results for control fibroblasts (NoC) and ciBAs (RoFB) have been previously
reported39. In this study, total RNA was prepared from 0.1 μM Ang II-treated ciBAs (RoFB + Ang II) derived from HDF38. The RNA integrity number values were more than 9 for all RNA samples
extracted using the FastGene RNA Premium Kit (Nippon Genetics, Tokyo, Japan). The library was prepared using the TruSeq stranded mRNA LT Sample Prep Kit (Illumina, San Diego, CA, USA),
following the manufacturer’s low-sample protocol. Paired-end sequencing (100 bp) was performed using the NovaSeq 6000 System (Illumina). Trimmed reads were mapped to a reference genome (NCBI
GRCh37) using HISAT2. After the read mapping, StringTie was used for transcript assembly. After the assembly, the abundance of genes/transcripts was calculated from read counts and
normalised as fragments per kilobase of transcript per million mapped sequence reads (FPKM). For the identification of DEGs, statistical analysis was performed using FC and an exact test
using edgeR per comparison pair. Significant results satisfying the conditions of |FC|≥ 2 and an exact test _p_-value < 0.05 were selected. If more than one read count value was zero, it
was excluded from the analysis. Heat maps were generated using Heatmapper (http://www.heatmapper.ca/)59. Hierarchical clustering analysis was performed based on the Euclidean distance. Each
row represents a gene, and each column represents the z-scored FPKM of each sample. The green and magenta gradients represent lower and higher gene expression, respectively. GO enrichment
analysis was performed using DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/)60. STATISTICAL ANALYSES All the results are presented as the mean ± standard deviation (SD).
Differences between two independent groups were evaluated by two-tailed Student’s t-test using the Excel (Microsoft, WA, USA) program. Differences between multiple independent groups were
analysed by one-way ANOVA or two-way ANOVA with Tukey’s multiple comparison test, unless otherwise stated in the figure legends, using an R-based statistical software, EZR version 1.65
(Saitama Medical Center, Jichi Medical University, Saitama, Japan)61. Statistical significance was defined as _p_ < 0.05. DATA AVAILABILITY The RNA-sequencing data have been deposited in
the DNA Data Bank of Japan (DDBJ) Sequenced Read Archive (https://www.ddbj.nig.ac.jp/dra/index-e.html) under the accession number DRR530882. REFERENCES * Lotta, L. A. _et al._ Association of
genetic variants related to gluteofemoral vs abdominal fat distribution with type 2 diabetes, coronary disease, and cardiovascular risk factors. _J. Am. Med. Assoc._ 320, 2553–2563 (2018).
Article CAS Google Scholar * Bienboire-Frosini, C. _et al._ The role of brown adipose tissue and energy metabolism in mammalian thermoregulation during the perinatal period. _Animals
(Basel)_ 13, 1–29 (2023). Google Scholar * Cohen, P. & Kajimura, S. The cellular and functional complexity of thermogenic fat. _Nat. Rev. Mol. Cell Biol._ 22, 393–409 (2021). Article
CAS PubMed PubMed Central Google Scholar * Fedorenko, A., Lishko, P. V. & Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. _Cell_ 151,
400–413 (2012). Article CAS PubMed PubMed Central Google Scholar * Cypess, A. M. Does activating brown fat contribute to important metabolic benefits in humans? Yes!. _J. Clin. Invest._
133, e175282 (2023). Article PubMed PubMed Central Google Scholar * Bornstein, M. R. _et al._ Comprehensive quantification of metabolic flux during acute cold stress in mice. _Cell
Metab._ 35, 2077-2092.e6 (2023). Article CAS PubMed Google Scholar * Becher, T. _et al._ Brown adipose tissue is associated with cardiometabolic health. _Nat. Med._ 27, 58–65 (2021).
Article CAS PubMed PubMed Central Google Scholar * Paul, M., Poyan Mehr, A. P. & Kreutz, R. Physiology of local renin-angiotensin systems. _Physiol. Rev._ 86, 747–803 (2006).
Article CAS PubMed Google Scholar * Faloia, E. _et al._ Comparison of circulating and local adipose tissue renin-angiotensin system in normotensive and hypertensive obese subjects. _J.
Endocrinol. Invest._ 25, 309–314 (2002). Article CAS PubMed Google Scholar * Engeli, S. _et al._ Weight loss and the renin-angiotensin-aldosterone system. _Hypertension_ 45, 356–362
(2005). Article CAS PubMed Google Scholar * Pahlavani, M., Kalupahana, N. S., Ramalingam, L. & Moustaid-Moussa, N. Regulation and functions of the renin-angiotensin system in white
and brown adipose tissue. _Compr. Physiol._ 7, 1137–1150 (2017). Article PubMed PubMed Central Google Scholar * Brücher, R., Cifuentes, M., Acuña, M. J., Albala, C. & Rojas, C. V.
Larger anti-adipogenic effect of angiotensin II on omental preadipose cells of obese humans. _Obesity (Silver Spring)_ 15, 1643–1646 (2007). Article PubMed Google Scholar * Matsushita,
K., Wu, Y., Okamoto, Y., Pratt, R. E. & Dzau, V. J. Local renin angiotensin expression regulates human mesenchymal stem cell differentiation to adipocytes. _Hypertension_ 48, 1095–1102
(2006). Article CAS PubMed Google Scholar * Than, A., Leow, M. K. S. & Chen, P. Control of adipogenesis by the autocrine interplays between angiotensin 1–7/Mas receptor and
angiotensin II/AT1 receptor signaling pathways. _J. Biol. Chem._ 288, 15520–15531 (2013). Article CAS PubMed PubMed Central Google Scholar * Fuentes, P., Acuña, M. J., Cifuentes, M.
& Rojas, C. V. The anti-adipogenic effect of angiotensin II on human preadipose cells involves ERK1,2 activation and PPARG phosphorylation. _J. Endocrinol._ 206, 75–83 (2010). Article
CAS PubMed Google Scholar * Kouyama, R. _et al._ Attenuation of diet-induced weight gain and adiposity through increased energy expenditure in mice lacking angiotensin II type 1a
receptor. _Endocrinology_ 146, 3481–3489 (2005). Article CAS PubMed Google Scholar * Li, A. _et al._ The gene knockout of angiotensin II type 1a receptor improves high-fat diet-induced
obesity in rat via promoting adipose lipolysis. _PLoS One_ 17, e0267331 (2022). Article CAS PubMed PubMed Central Google Scholar * Frigolet, M. E., Torres, N. & Tovar, A. R. The
renin-angiotensin system in adipose tissue and its metabolic consequences during obesity. _J. Nutr. Biochem._ 24, 2003–2015 (2013). Article CAS PubMed Google Scholar * Kalupahana, N. S.
& Moustaid-Moussa, N. The renin-angiotensin system: A link between obesity, inflammation and insulin resistance. _Obes. Rev._ 13, 136–149 (2012). Article CAS PubMed Google Scholar *
Schorr, U., Blaschke, K., Turan, S., Distler, A. & Sharma, A. M. Relationship between angiotensinogen, leptin and blood pressure levels in young normotensive men. _J. Hypertens._ 16,
1475–1480 (1998). Article CAS PubMed Google Scholar * Kim, J. Y. _et al._ The relationship between cold exposure and hypertension. _J. Occup. Health_ 45, 300–306 (2003). Article PubMed
Google Scholar * Brook, R. D., Weder, A. B. & Rajagopalan, S. ‘Environmental Hypertensionology’ the effects of environmental factors on blood pressure in clinical practice and
research. _J. Clin. Hypertens. (Greenwich)_ 13, 836–842 (2011). Article PubMed Google Scholar * Wang, X., Sun, Z. & Cade, R. Prolonged attenuation of cold-induced hypertension by
adenoviral delivery of renin antisense. _Kidney Int._ 68, 680–687 (2005). Article CAS PubMed Google Scholar * Sun, Z. Cardiovascular responses to cold exposure. _Front. Biosci. (Elite
Ed.)_ 2, 495–503 (2010). Article PubMed Google Scholar * Sun, Z., Cade, R., Katovich, M. J. & Fregly, M. J. Body fluid distribution in rats with cold-induced hypertension. _Physiol.
Behav._ 65, 879–884 (1999). Article CAS PubMed Google Scholar * Barney, C. C., Katovich, M. J., Fregly, M. J. & Tyler, P. E. Changes in beta-adrenergic responsiveness of rats during
chronic cold exposure. _J. Appl. Physiol. Respir. Environ. Exerc. Physiol._ 49, 923–929 (1980). CAS PubMed Google Scholar * Fregly, M. J., Field, F. P., Nelson, E. L., Tyler, P. E. &
Dasler, R. Effect of chronic exposure to cold on some responses to catecholamines. _J. Appl. Physiol. Respir. Environ. Exerc. Physiol._ 42, 349–354 (1977). CAS PubMed Google Scholar *
Sun, Z., Cade, R. & Morales, C. Role of central angiotensin II receptors in cold-induced hypertension. _Am. J. Hypertens._ 15, 85–92 (2002). Article CAS PubMed Google Scholar * Sun,
Z., Wang, X., Wood, C. E. & Cade, J. R. Genetic AT1A receptor deficiency attenuates cold-induced hypertension. _Am. J. Physiol. Regul. Integr. Comp. Physiol._ 288, R433–R439 (2005).
Article CAS PubMed Google Scholar * Cassis, L. _et al._ Cold exposure regulates the renin-angiotensin system. _J. Pharmacol. Exp. Ther._ 286, 718–726 (1998). CAS PubMed Google Scholar
* Luo, B., Zhang, S., Ma, S., Zhou, J. & Wang, B. Effects of cold air on cardiovascular disease risk factors in rat. _Int. J. Environ. Res. Public Health_ 9, 2312–2325 (2012). Article
PubMed PubMed Central Google Scholar * Zhang, X., Zhang, S., Wang, C., Wang, B. & Guo, P. Effects of moderate strength cold air exposure on blood pressure and biochemical indicators
among cardiovascular and cerebrovascular patients. _Int. J. Environ. Res. Public Health_ 11, 2472–2487 (2014). Article PubMed PubMed Central Google Scholar * Cassis, L. A. Role of
angiotensin II in brown adipose thermogenesis during cold acclimation. _Am. J. Physiol._ 265, E860–E865 (1993). CAS PubMed Google Scholar * King, V. L., English, V. L., Bharadwaj, K.
& Cassis, L. A. Angiotensin II stimulates sympathetic neurotransmission to adipose tissue. _Physiol. Rep._ 1, e00014 (2013). Article PubMed PubMed Central Google Scholar * Cassis, L.
A. & Dwoskin, L. P. Presynaptic modulation of neurotransmitter release by endogenous angiotensin II in brown adipose tissue. _J. Neural Transm. Suppl._ 34, 129–137 (1991). CAS PubMed
Google Scholar * Tuo, B. X. _et al._ Analysis of differentially expressed genes in cold-exposed mice to investigate the potential causes of cold-induced hypertension. _Exp. Ther. Med._ 8,
110–114 (2014). Article PubMed PubMed Central Google Scholar * Takeda, Y., Harada, Y., Yoshikawa, T. & Dai, P. Direct conversion of human fibroblasts to brown adipocytes by small
chemical compounds. _Sci. Rep._ 7, 4304 (2017). Article ADS PubMed PubMed Central Google Scholar * Takeda, Y. & Dai, P. A developed serum-free medium and an optimized chemical
cocktail for direct conversion of human dermal fibroblasts into brown adipocytes. _Sci. Rep._ 10, 3775 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Takeda, Y.,
Yoshikawa, T. & Dai, P. Transcriptome analysis reveals brown adipogenic reprogramming in chemical compound-induced brown adipocytes converted from human dermal fibroblasts. _Sci. Rep._
11, 5061 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Takeda, Y. & Dai, P. Chronic fatty acid depletion induces uncoupling protein 1 (UCP1) expression to
coordinate mitochondrial inducible proton leak in a human-brown-adipocyte model. _Cells_ 11, 2038 (2022). Article CAS PubMed PubMed Central Google Scholar * Takeda, Y., Harada, Y.,
Yoshikawa, T. & Dai, P. Mitochondrial energy metabolism in the regulation of thermogenic brown fats and human metabolic diseases. _Int. J. Mol. Sci._ 24, 1352 (2023). Article CAS
PubMed PubMed Central Google Scholar * Morimoto, H. _et al._ Angiotensin 1–7 stimulates brown adipose tissue and reduces diet-induced obesity. _Am. J. Physiol. Endocrinol. Metab._ 314,
E131–E138 (2018). Article PubMed Google Scholar * Cao, X. _et al._ ACE2 pathway regulates thermogenesis and energy metabolism. _eLife_ 11, e72266 (2022). Article CAS PubMed PubMed
Central Google Scholar * Evangelista, F. S. & Bartness, T. J. Central antgiotensin 1–7 triggers brown fat thermogenesis. _Physiol. Rep._ 11, e15621 (2023). Article CAS PubMed PubMed
Central Google Scholar * Blondin, D. P. _et al._ Human brown adipocyte thermogenesis is driven by β2-AR stimulation. _Cell Metab._ 32, 287-300.e7 (2020). Article CAS PubMed Google
Scholar * Mehta, P. K. & Griendling, K. K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. _Am. J. Physiol. Cell Physiol._ 292,
C82–C97 (2007). Article CAS PubMed Google Scholar * Forrester, S. J. _et al._ Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. _Physiol.
Rev._ 98, 1627–1738 (2018). Article CAS PubMed PubMed Central Google Scholar * Valente, V. _et al._ Modulation of insulin resistance by renin angiotensin system inhibitors: Implications
for cardiovascular prevention. _Monaldi Arch. Chest Dis._ https://doi.org/10.4081/monaldi.2021.1602 (2021). Article PubMed Google Scholar * Kalupahana, N. S. _et al._ Overproduction of
angiotensinogen from adipose tissue induces adipose inflammation, glucose intolerance, and insulin resistance. _Obesity (Silver Spring)_ 20, 48–56 (2012). Article CAS PubMed Google
Scholar * Sechi, L. A. _et al._ Effects of angiotensin II on insulin receptor binding and mRNA levels in normal and diabetic rats. _Diabetologia_ 40, 770–777 (1997). Article CAS PubMed
Google Scholar * Baba, T., Drenckhan, M., Neugebauer, S. & Klein, H. Effect of angiotensin II and bradykinin on insulin-stimulated tyrosine kinase activity of insulin receptor.
_Diabetologia_ 41, 741–742 (1998). Article CAS PubMed Google Scholar * Andreozzi, F., Laratta, E., Sciacqua, A., Perticone, F. & Sesti, G. Angiotensin II impairs the insulin
signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser312 and Ser616 in human umbilical vein endothelial cells. _Circ.
Res._ 94, 1211–1218 (2004). Article CAS PubMed Google Scholar * Kim, J. A., Jang, H. J., Martinez-Lemus, L. A. & Sowers, J. R. Activation of mTOR/p70S6 kinase by ANG II inhibits
insulin-stimulated endothelial nitric oxide synthase and vasodilation. _Am. J. Physiol. Endocrinol. Metab._ 302, E201–E208 (2012). Article CAS PubMed Google Scholar * Goossens, G. H.,
Blaak, E. E., Arner, P., Saris, W. H. M. & van Baak, M. A. Angiotensin II: A hormone that affects lipid metabolism in adipose tissue. _Int. J. Obes. (Lond)_ 31, 382–384 (2007). Article
CAS PubMed Google Scholar * Frantz, E. D. C. _et al._ Modulation of the renin–angiotensin system in white adipose tissue and skeletal muscle: Focus on exercise training. _Clin. Sci.
(Lond)_ 132, 1487–1507 (2018). Article ADS CAS PubMed Google Scholar * Abbas, N. A. T., Fayed, F. A., El Sebaey, R. S. & Hassan, H. A. Telmisartan and candesartan promote browning
of white adipose tissue and reverse fatty liver changes in high fat diet fed male albino rats. _Naunyn Schmiedebergs Arch. Pharmacol._ 397(4), 2359–2378 (2023). Article PubMed Google
Scholar * Takeda, Y. & Dai, P. Capsaicin directly promotes adipocyte browning in the chemical compound-induced brown adipocytes converted from human dermal fibroblasts. _Sci. Rep._ 12,
6612 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Jing, R. _et al._ A screen using iPSC-derived hepatocytes reveals NAD+ as a potential treatment for mtDNA depletion
syndrome. _Cell Rep._ 25, 1469-1484.e5 (2018). Article CAS PubMed PubMed Central Google Scholar * Babicki, S. _et al._ Heatmapper: Web-enabled heat mapping for all. _Nucleic Acids Res._
44, W147–W153 (2016). Article CAS PubMed PubMed Central Google Scholar * Sherman, B. T. _et al._ David: A web server for functional enrichment analysis and functional annotation of
gene lists (2021 update). _Nucleic Acids Res._ 50, W216–W221 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Kanda, Y. Investigation of the freely available easy-to-use
software ‘EZR’ for medical statistics. _Bone Marrow Transplant_ 48, 452–458 (2013). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This research was supported by
the Kataoka Corporation (Kyoto, Japan), JSPS KAKENHI Grant Numbers JP21K12669 and JP24K03268, Takeda Science Foundation, Lotte Research Promotion Grant, and Suzuken Memorial Foundation.
AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Cellular Regenerative Medicine, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, 465 Kajii-cho,
Kawaramachi-Hirokoji, Kamigyo-ku, Kyoto, 602-8566, Japan Yukimasa Takeda & Ping Dai * Louis Pasteur Center for Medical Research, 103-5 Tanaka-Monzen-cho, Sakyo-ku, Kyoto, 606-8225, Japan
Toshikazu Yoshikawa * Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, 465 Kajii-cho,
Kawaramachi-Hirokoji, Kamigyo-ku, Kyoto, 602-8566, Japan Toshikazu Yoshikawa Authors * Yukimasa Takeda View author publications You can also search for this author inPubMed Google Scholar *
Toshikazu Yoshikawa View author publications You can also search for this author inPubMed Google Scholar * Ping Dai View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS Y.T. and P.D. designed the experiments, Y.T. conducted the experiments, P.D. and T.Y. provided comments and conceptual advice, and Y.T. wrote the manuscript. All
the authors reviewed the manuscript. CORRESPONDING AUTHORS Correspondence to Yukimasa Takeda or Ping Dai. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests.
ADDITIONAL INFORMATION PUBLISHER'S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION
SUPPLEMENTARY INFORMATION. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise
in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the
permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Takeda, Y., Yoshikawa, T. & Dai, P. Angiotensin II participates in mitochondrial thermogenic functions via the activation of glycolysis
in chemically induced human brown adipocytes. _Sci Rep_ 14, 10789 (2024). https://doi.org/10.1038/s41598-024-61774-0 Download citation * Received: 22 February 2024 * Accepted: 09 May 2024 *
Published: 11 May 2024 * DOI: https://doi.org/10.1038/s41598-024-61774-0 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * Brown adipocytes *
Angiotensin II * UCP1 * Mitochondria * Adaptive thermogenesis * Glycolysis