Mitochondrial dysfunction impairs osteogenesis, increases osteoclast activity, and accelerates age related bone loss
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The pathogenesis of declining bone mineral density, a universal feature of ageing, is not fully understood. Somatic mitochondrial DNA (mtDNA) mutations accumulate with age in human
tissues and mounting evidence suggests that they may be integral to the ageing process. To explore the potential effects of mtDNA mutations on bone biology, we compared bone
microarchitecture and turnover in an ageing series of wild type mice with that of the _PolgA__mut/mut_ mitochondrial DNA ‘mutator’ mouse. In vivo analyses showed an age-related loss of bone
in both groups of mice; however, it was significantly accelerated in the _PolgA__mut/mut_ mice. This accelerated rate of bone loss is associated with significantly reduced bone formation
rate, reduced osteoblast population densities, increased osteoclast population densities, and mitochondrial respiratory chain deficiency in osteoblasts and osteoclasts in _PolgA__mut/mut_
mice compared with wild-type mice. In vitro assays demonstrated severely impaired mineralised matrix formation and increased osteoclast resorption by _PolgA__mut/mut_ cells. Finally,
application of an exercise intervention to a subset of _PolgA__mut/mut_ mice showed no effect on bone mass or mineralised matrix formation in vitro. Our data demonstrate that mitochondrial
dysfunction, a universal feature of human ageing, impairs osteogenesis and is associated with accelerated bone loss. SIMILAR CONTENT BEING VIEWED BY OTHERS PREMATURE AGING OF SKELETAL
STEM/PROGENITOR CELLS RATHER THAN OSTEOBLASTS CAUSES BONE LOSS WITH DECREASED MECHANOSENSATION Article Open access 05 July 2023 DELETION OF SKELETAL MUSCLE _AKT1_/_2_ CAUSES OSTEOSARCOPENIA
AND REDUCES LIFESPAN IN MICE Article Open access 05 October 2022 LOSS OF NOTCH SIGNALING IN SKELETAL STEM CELLS ENHANCES BONE FORMATION WITH AGING Article Open access 27 September 2023
INTRODUCTION Normal bone homeostasis requires maintenance of a delicate balance between the continuous processes of bone resorption by osteoclasts and new bone formation by osteoblasts1,2.
Following attainment of peak bone mass in early adulthood, a decline in bone mineral density (BMD) ensues and continues unabated for the remainder of life in both humans and mice3,4.
Progressive deterioration in bone microarchitecture with declining mineralisation levels and increasing porosity occurs, frequently culminating in osteoporosis5,6,7. The process affects
males and females universally, with a transiently accelerated rate of loss observed in the latter secondary to menopausal oestrogen loss in humans3,8. Diminishing bone strength is inherent
to declining BMD levels, the consequence of which is increasing risk of fragility fractures occurring with increasing age9,10. The pathogenesis of bone loss is not fully understood with
various theories postulated such as increasing secretion of endogenous glucocorticoids11,12 and decreasing levels of sex hormones13,14,15,16, physical activity levels17,18,19 and insulin
like growth factor I20,21. However, intracellular changes within bone tissue that occur with age, specifically accumulating mitochondrial DNA (mtDNA) mutations, may play a significant role
in the failure of bone homeostasis leading to declining BMD levels. With age, somatic mtDNA mutations accumulate in post mitotic tissues such a brain and muscle22,23,24,25, and mitotic
tissue such as gut26,27. Mounting evidence in recent years, particularly that provided by animal models, has suggested that these may be intrinsic to the ageing process28,29,30. One of the
most important functions of mitochondria in any cell is the production of energy in the form of adenosine triphosphate (ATP) via the process of oxidative phosphorylation31, although some ATP
production can occur via much less efficient pathways such as glycolysis. For normal cellular function to occur, a minimum energy requirement must exist32. Cellular dysfunction has been
shown to occur once genomes containing mtDNA mutations outnumber the presence of normal mtDNA sufficiently, depending on the type of mutations present, a phenomenon known as the threshold
effect33,34. mtDNA polymerase gamma (Polg) is the only DNA polymerase found in mitochondria and is responsible for replication and repair of mtDNA within all cell types35. The
_PolgA__mut/mut_ mitochondrial ‘mutator’ mouse possesses a defective version of the only proof reading domain of mtDNA polymerase36 which causes it to accumulate mitochondrial DNA point
mutations at 3–5 times the rate of wild type mice resulting in a premature ageing phenotype, making it an excellent mouse model of ageing37. One of the most prominent consequences of this
prematurely ageing phenotype is osteoporosis. Further evidence for mitochondrial dysfunction as a potential contributor to osteoporosis is seen in mice with a mitochondrial transcription
factor A (TFAM) knockout specific to osteoclasts, the result of which is increased resorption when grown on dentine compared to normal osteoclasts38. Mice with global and osteocyte specific
knockdown of superoxide dismutase (Sod2), an enzyme which protects against mitochondrial oxidative stress, also develop osteoporosis prematurely39. Previous work studying the effects of
exercise on _PolgA__mut/mut_ mice, showed that regular exercise over a 5 month period ameliorated the increased rate of mtDNA mutations, increased mtDNA copy number and completely abolished
the accelerated ageing phenotype, at 8 months of age. However, the effects of exercise on accelerated bone depletion was not evaluated40. Similarly, endurance exercise in human subjects has
been shown to increase oxidative capacity of mitochondria within muscle, although the effects on bone mass are unknown41,42. The normal function of osteoblasts in producing bone entails the
production of a collagen framework and its subsequent mineralisation43. The susceptibility of this cell line to mtDNA mutations was previously unknown. We developed a quadruple
immunofluorescence assay which accurately quantifies respiratory chain protein expression in bone cells, and demonstrated that mitochondrial COX-I and NDUFB8 protein expression in bone
lining osteoblasts decreases with age in wild type mice (4 months vs. 11 months old), and is significantly reduced in _PolgA__mut/mut_ mice aged 11 months compared to age matched wild type
controls44. In order to evaluate the potential effects of osteoblast and osteoclast mitochondrial dysfunction on bone homeostasis, here we have assessed the effects of the _PolgA__mut/mut_
genotype on bone phenotype, capacity of osteogenic cell lines to form mineralised matrix in vitro_,_ osteoclast respiratory protein expression and resorption capacity in vitro_._ The effects
of exercise on bone phenotype and in vitro mineralisation are also presented. METHODS MICE Mitochondrial mutator mice (_PolgA__mut/mut_) were generated that had a knock-in missense mutation
(D257A) in the second endonuclease proofreading domain of the _PolgA_ catalytic subunit of the mtDNA polymerase, using a C57BL/6 background mouse28. Female and male mice were used, with
comparisons made between _PolgA__mut/mut_ and wild type controls from the previous generation to ensure no transmission of mtDNA mutations to ‘wild-type’ mice through the maternal line45.
Animals were housed in single sex cages and cared for in compliance with the Animals (Scientific Procedures) Act 1984 in a purpose built facility. There was no variance in housing
conditions, feed or water provision between _PolgA__mut/mut_ and wild type littermate controls. All animal experiments were approved by and conducted in compliance with the UK Home Office
(PPL P3052AD70) and the Newcastle University Animal Welfare Ethical Review Board (AWERB 425). ASSESSMENT OF VOLUMETRIC BONE (BV/TV) Lumbar spines and femurs were extracted from mice aged 4,
7 and 11 months and fixed for 72 h in 10% normal buffered formalin (NBF) after removal of soft tissues. Micro computed tomography (CT) scans of the lumbar spines and distal femur were then
performed to determine BV/TV. Imaging was performed using a Skyscan 1,272 micro CT scanner. A beam intensity of 50Kv and 200µA was used with a 0.5 ml Al filter. A resolution of 4.3 µm and
specimen rotation angle of 0.3° was used for femoral trabecular scans and a resolution of 8.6 µm and specimen rotation angle of 0.5 was used for femoral cortical scans. For lumbar vertebrae
assessment, the L5 vertebrae was assessed, not including cortical bone or end plates. A resolution of 4.5 µm and a specimen rotation angle of 0.4 was used. Volumetric bone mass was recorded
as a measure of bone volume (BV)/tissue volume (TV). Trabecular thickness, trabecular separation and trabecular number were also recorded in femoral and lumbar vertebrae trabecular bone.
Cortical thickness was measured in diaphyseal femoral cortical bone. BONE HISTOMORPHOMETRY Tibiae from female mice aged 7 months were dissected, fixed in 4% formaldehyde for 24 h and
embedded in methyl methacrylate without decalcification. 5 μm sections were taken for bone histomorphometry, performed as detailed previously46. In brief, sections were stained with Von
Kossa stain to analyse bone mineralisation, 1% Toluidine blue to analyse osteoblast number, and stained for tartrate-resistant acid phosphatase (TRAcP) to analyse osteoclast number and
surface area. To study the dynamic properties of bone, mice were injected intra-peritoneally 10-days and 2-days prior to sacrifice with 30 mg/kg alizarin complexone and 20 mg/kg calcein in a
2% sodium bicarbonate pH 7.4 solution, respectively. Trabecular bone histomorphometric measurements were taken from fluorescent images of 5 μm sections acquired using a Zeiss Axio Imager 2
microscope and were used to calculate the bone formation rate (BFR). All bone histomorphometry analyses were performed on matched sections from 3 mice per genotype and were performed on the
trabecular bone in the proximal tibia, 100 μm below the physis. IN VITRO ASSESSMENT OF MINERALISED MATRIX FORMATION BY OSTEOGENIC CELLS Cells from mice aged 4, 7 and 11 months were compared.
Following dissection of mouse femurs, the epiphyses were cut and the bone marrow from individual femurs was harvested by flushing the intramedullary canal with DMEM using a 25G needle. Each
sample was centrifuged at 300× _g_ and the supernatant removed, before the cell pellet was suspended in a T25 flask in 5mls of alpha modified minimum essential media (MEM). Alpha MEM was
supplemented with 10% FBS, 2 mM L-glutamine, penicillin (100 u/ml), streptomycin (100 µg/ml) and amphotericin (0.25 µg/ml). After 24 h of incubation at 37 °C non-adherent cells were removed,
aided by PBS wash steps, following which fresh media was applied. Media was changed again on day 6 and cells were harvested for use in mineralisation assays on day 10. At this point, cells
were washed with PBS and dissociated using TrypLE express. Cells from each mouse were then plated at a density of 6.5 × 104 in all wells of a 12 well plate in osteogenic media (alpha MEM
supplemented as above with the addition of 50 µg/ml sodium ascorbate and 2 mM beta-glycerophosphate47. A full media change was performed every 3 days for 21 days at which point the cells
were fixed briefly in 4% PFA. 7 of the wells were washed in 70% ethanol, air dried, stained with 2% alizarin red (which binds to calcium within mineralised bone) in dH2O for 10 min, washed 3
times with 50% ethanol and air dried. The 5 remaining wells were washed with TBS prior to ALP staining which was performed by combining 10 mg napthol MX (dissolved in 400 µl
N,N-Dimethylformamide), with 60 mg of fast blue RR salt (Sigma F0500) in 100mls of tris-buffered saline (TBS), at a pH of 8.2. Cells were incubated in the dark for 30 min in this solution,
following which, nuclei of cells within the same 5 wells were stained with Hoechst. As per previous work, we confirmed osteogenic differentiation in vitro by staining for ALP
activity47,48,49,50,51,52,53. ALP is a phospho-ester, imperative to the mineralisation process54,55. Fluorescent microscopy (405 nm) was used to image Hoechst stained cells, with 15 images
taken from each of 5 wells at 4× magnification. Matlab automated software was used to record cell counts from these images. 26 colour brightfield images were taken of all alizarin red and
ALP stained wells at 4× magnification. Image analysis was performed using an RGB filter within Volocity (PerkinElmer, Coventry, UK) software to detect alizarin red staining and the purple of
ALP staining. The average surface area of mineralisation as depicted by alizarin red staining was recorded for each mouse cell line, as was the surface area and number of osteogenic cells,
identified by positive ALP staining. Final results were expressed as average surface area of mineralisation per average area of ALP stained cells. To calculate population density of
osteogenic cells in each image, the ratio of ALP positive cells to total cell count per image (as identified by Hoescht nuclear staining) was also recorded for each cell line. EFFECTS OF
EXERCISE ON BONE AND PHENOTYPE AND IN VITRO MINERALISATION A subset of male _PolgA__mut/mut_ mice were exercised from the age of 16 weeks, for 4 days per week using a treadmill. Following an
initial 10-week acclimatisation period of gradually increasing intensity, the final protocol consisted of the mice running at 12 cm/s for 5 min, 20 cm/s for 40 min, and finally for 12 cm/s
for 5 min on each of the 4 days. Tissue was harvested from mice aged 11 months to study whether exercise had any effects on bone density and functional capacity of osteogenic cells, in
comparison to non-exercised _PolgA__mut/mut_ mice aged 11 months. IN VITRO ASSESSMENT OF OSTEOCLAST FUNCTION Osteoclasts from male _PolgA__mut/mut_ and wild type litter mates, aged 11 months
were compared using a previously published method56. On day 1, following dissection of mouse femurs, the epiphyses were cut and the bone marrow from individual femurs was harvested by
flushing the intramedullary canal with DMEM using a 25G needle. Each sample was centrifuged at 300× _g_ and the supernatant removed, before the cell pellet was suspended in a T25 flask in
5mls of alpha modified minimum essential media (MEM). Alpha-MEM was supplemented with 10% FBS, 2 mM L-glutamine, penicillin (100u/ml), streptomycin (100 µg/ml) and amphotericin (0.25 µg/ml),
10−7 M prostaglandin E2 and 2.5 ng/ml of M-CSF. Cells were incubated for 24 h at 37 °C in an incubator supplemented with 5% CO2. On day 2, non-adherent cells from each flask were removed,
aided by PBS wash steps. These were centrifuged at 300× _g_ for 5 min and the supernatant removed. The cell pellet was then suspended in alpha MEM at a density of 5 × 106 cells/ml. Alpha-MEM
was supplemented at this stage and for the remainder of the experiment with 10% FBS, 2 mM L-glutamine, penicillin (100 u/ml), streptomycin (100 µg/ml), amphotericin (0.25 µg/ml), 10-7 M
prostaglandin E2, 10 ng/ml M-CSF and 3 ng/ml of mouse RANKL. Using 96-well plates, cells from each mouse were seeded on to four 0.5 mm dentine discs (1 × 106 cells per disc in 200 µl media.
Dentine was derived from walrus tusk (Oxford Biosystems), and had been pre-soaked in PBS. Following a further 24 h, on day 3, discs were transferred to 6 well plates, with 2 mls of fresh
supplemented alpha MEM in each well. A 50% media change was carried out on day 5. On day 7, a full media change was performed with the media having been acidified with the addition of HCl to
reduce the pH to 6.9 (at incubator conditions) to activate osteoclast resorption. After 24 h in acidified media, dentine discs were washed in PBS and fixed using 2% glutaraldehyde in
Sorensen’s solution, for 5 min. TRAP staining was performed to detect osteoclasts. Osteoclasts were defined as TRAP positive, multi-nucleated cells. TRAP staining solution, pH 5.0, contained
0.1 M sodium acetate, 7.5 mM L-( +) tartaric acid, 0.2 mM naphthol AS-MX and 1.5 mM fast red violet LB salt (Sigma Aldrich). Cells were stained at 37 °C for 30 min. Transmitted light
microscopy at 10× optical magnification was used to record images from 10 different areas of each disc. NIS-elements advanced research software (Nikon) was used to identify multinucleated
TRAP positive osteoclasts to derive an average number of cells per mm2. Following osteoclast cell counts, cells were removed from the dentine discs using cotton buds and 0.25 M ammonium
hydroxide. The dentine discs were then stained by immersion in 1% toluidine blue dissolved in 0.1 M sodium tetraborate for 3 min. Discs were then rinsed in water to remove excess staining.
Transmitted light microscopy was used at 10 × optical magnification to take 10 images from different areas of each disc. Using NIS-elements advanced research software (Nikon), average values
were obtained for number of pits/mm2 and average pit size (µm) for each disc. IN VIVO ASSESSMENT OF OSTEOBLAST AND OSTEOCLAST MITOCHONDRIAL RESPIRATORY CHAIN PROTEIN EXPRESSION Quadruple
immunofluorescence was performed to quantify levels of mitochondrial proteins in osteoblasts and osteoclasts as previously described44. Comparisons were made between wild type mice aged 4
and 11 months, and _PolgA__mut/mut_ mice aged 11 months. Data from 4-month-old wild type animals were used as the reference control. An antibody to cathepsin K was used to detect
osteoclasts, and respiratory chain complex subunits within were detected using mouse monoclonal primary antibodies against NADH dehydrogenase [ubiquinone] I beta subcomplex subunit 8
(NDUFB8) and cytochrome _c _oxidase (COX) subunit 1 (COXI). These were applied in combination with a monoclonal antibody against the outer mitochondrial membrane protein porin (VDAC1). Porin
is a nuclear-encoded, voltage gated ion channel present in abundance in the mitochondrial membrane, and its presence serves as a marker for mitochondrial mass57. NDUFB8 is a nuclear-DNA
encoded subunit of Complex I58 and COX-I is a mtDNA-encoded subunit of Complex IV59. Imaris image analysis software (Bitplane, v.8.4) was used to detect osteoclasts (546 nm) and areas within
these that were positive for the mitochondrial mass marker porin (405 nm). For each of these areas of intracellular mitochondrial staining, the software provided average signal intensity
values for porin (405 nm), COX-I/MTCO1 (488 nm) and Complex I/NDUFB8 (647 nm) within each cell. STATISTICAL ANALYSIS Prism (GraphPad) was used for all statistical analysis of micro CT scan,
histomorphometry and cell culture data. Rstudio was used for analysis of osteoclast immunofluorescence data. RESULTS MICRO-CT SCANNING OF FEMURS AND LUMBAR SPINES DEMONSTRATES ACCELERATED
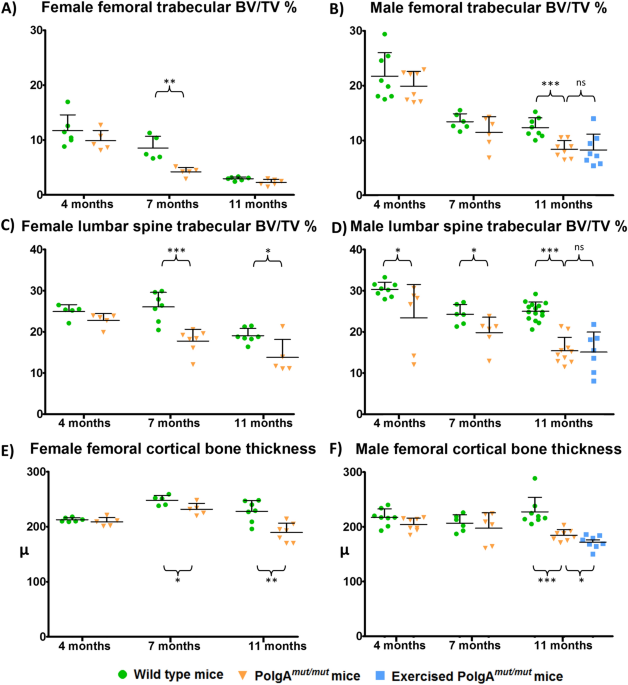
BONE LOSS IN _POLGA_ _MUT/MUT_ MICE Bone volume/tissue volume (BV/TV) of extracted femurs was evaluated using micro-CT scanning (Fig. 1) and unpaired 2-tailed t tests for comparisons. The
femoral BV/TV decreased at an accelerated rate in _PolgA__mut/mut_ mice compared to age and sex matched wild type controls with a significantly reduced trabecular BV/TV in both female and
male _PolgA__mut/mut_ mice by the ages of 7 and 11 months respectively (_p_ = 0.003 and < 0.0001). The same comparisons (Supplemental Data S1) also show reduced levels of trabecular bone
thickness, increased trabecular separation and reduced trabecular number in 11 month old _PolgA__mut/mut_ males (_p_ = 0.0009, 0.007 and 0.025 respectively) and in 7 month old
_PolgA__mut/mut_ females (_p_ = 0.251, < 0.0001 and 0.004 respectively). A decrease in trabecular BV/TV of male and female wild type mice, with increasing age is also observed. Female
wild type mice demonstrate significantly reduced BV/TV compared to age matched wild type males at all 3 ages studied (_p_ < 0.001). Assessment of cortical bone thickness (Fig. 1 and
Supplemental Data S1) showed that it continued to increase in wild type mice between the ages of 4 and 7 months in females, in keeping with previous work40. However, significantly reduced
cortical thickness was seen in _PolgA__mut/mut_ female mice at 7 months (_p_ = 0.032), and at 11 months of age (_p_ = 0.005). Male _PolgA__mut/mut_ mice exhibited significantly reduced
cortical thickness at 11 months of age (_p_ = 0.0008). Trabecular bone from extracted lumbar vertebrae (L5) was also assessed (Fig. 1 and Supplemental Data S2). Compared with wild-type
controls, a significantly reduced BV/TV was observed in _PolgA__mut/mut_ males at 4, 7 and 11 months of age (_p_ = 0.035, 0.033, and < 0.0001 respectively), and in _PolgA__mut/mut_ female
mice at 7 and 11 months of age (_p_ = 0.0004 and 0.007 respectively). This corresponded with reduced trabecular thickness at 7 and 11 months in _PolgA__mut/mut_ males (_p_ = 0.016 and 0.002
respectively) and _PolgA__mut/mut_ females (_p_ = 0.040 and 0.0001 respectively). In _PolgA__mut/mut_ males, increased trabecular separation was observed at 4, 7 and 11 months (_p_ = 0.005,
0.027 and 0.003 respectively), with reduced trabecular number at 4 and 11 months (_p_ = 0.033 and < 0.0001 respectively). _PolgA__mut/mut_ females exhibited increased trabecular
separation and reduced trabecular number at 7 months (_p_ = 0.013 and < 0.0001 respectively). Again, exercise was not associated with any difference in trabecular bone mass observed in
_PolgA__mut/mut_ male mice at 11 months. Comparison of exercised and non-exercised male _PolgA__mut/mut_ mice aged 11 months demonstrated that exercise did not affect any of the studied
parameters on micro CT scan of whole femurs (trabecular BV/TV, trabecular thickness, trabecular separation, trabecular number) or lumbar vertebrae (trabecular BV/TV, trabecular thickness,
trabecular separation, trabecular number). However, a reduction in cortical thickness was observed in exercised _PolgA__mut/mut_ mice (_p_ = 0.038). HISTOMORPHOMETRY SHOWS REDUCED BV/TV,
REDUCED BONE FORMATION RATE, REDUCED OSTEOBLAST POPULATION DENSITY, AND INCREASED OSTEOCLAST POPULATION DENSITY IN _POLGA_ _MUT/MUT_ MICE The extracted tibiae of age matched wild type and
_PolgA__mut/mut_ mice were compared (unpaired 2 tailed t-tests). BV/TV and mineralisation (osteoid surface/bone surface) were found to be significantly lower in _PolgA__mut/mut_ mice (_p_ =
0.017 and 0.010 respectively) compared to age matched wild type mice. Analysis of mineralisation fronts following staged injection of alizarin complexone and calcein demonstrated
significantly reduced bone formation rates (_p_ = 0.0088) in _PolgA__mut/mut_ mice. The in vivo population densities of osteoblasts in _PolgA__mut/mut_ mice was found to be significantly
lower (_p_ = 0.005), but conversely, that of osteoclasts was significantly (_p_ = 0.017) higher (Fig. 2). FUNCTIONAL CAPACITY OF OSTEOGENIC CELLS TO PRODUCE MINERALISED MATRIX IN VITRO IS
REDUCED IN _POLGA_ _MUT/MUT_ CELLS Mineralisation assays were performed using bulk unfractionated skeletal mesenchymal cells. The average surface area of mineralised matrix formed and
corresponding surface area occupied by cells expressing ALP were recorded (Supplemental Data S3) for each cell line. Wild type osteogenic cells consistently produced mineralised matrix in
abundance as demonstrated by increased alizarin red staining. The ratio of mineralised matrix surface area to surface area occupied by ALP positive cells (Fig. 3) fell with increasing age in
the wild type mice, with significant reduction in mineralised matrix formation by 7 and 11 months in comparison to that at 4 months in both sexes (one way ANOVA, Bonferroni _p_ < 0.001).
Osteogenic cells differentiated from cells harvested from male and female _PolgA__mut/mut_ mice aged 4, 7 and 11 months all produced significantly less mineralised matrix in comparison to
age and sex matched wild type controls (_p_ < 0.0001, unpaired 2 tailed t-test). The exercise intervention made no significant difference to the capacity of osteogenic cells extracted
from _PolgA__mut/mut_ mice aged 11 months to produce mineralised matrix in vitro_._ No significant difference was observed in propensity for cell differentiation to ALP expressing osteogenic
cells in this cell line either. PROPENSITY OF OSTEOGENIC CELL DIFFERENTIATION TO CELLS EXPRESSING ALP IS UNAFFECTED IN MSCS HARVESTED FROM _POLGA_ _MUT/MUT_ MICE The population density of
ALP positive cells derived from bulk unfractionated skeletal mesenchymal cell harvest was examined by correlating the ratio of number of ALP positive cells to the number of all cell types
(identified by nuclear Hoescht staining) for each mouse of origin (Fig. 4). One-way ANOVA and Bonferroni post-test comparisons showed no significant difference between wild type and
_PolgA__mut/mut_ cell lines in their propensity for osteogenic cell differentiation in male and female cell lines at all 3 ages. Significant variation in final population density of ALP
positive cells is evident in age/sex matched groups with no significant positive correlation (Pearson) found in any age/sex matched cell line between the number of ALP positive cells formed
and total number of cells present as identified by Hoechst nuclear staining (Supplemental Data S4). OSTEOCLAST IN VITRO RESORPTION ASSAYS DEMONSTRATE INCREASED OSTEOCLAST ACTIVITY Following
monocyte extraction from mice aged 11 months, and osteoclast culture on dentine discs, cells were removed and resorption pits were studied. Unpaired 2 tailed t-tests were performed for all
comparisons. The resorption pits formed by wild type osteoclasts were larger than those formed by _PolgA__mut/mut_ osteoclasts with average sizes of 534 µm and 333 µm respectively (_p_ =
0.0008). The ratio of resorption pits to osteoclasts was analysed (Fig. 5). The ratio of pits to osteoclasts was significantly higher following _PolgA__mut/mut_ osteoclast culture, with 9.3
pits observed per osteoclast compared to only 3.2 pits per osteoclast following wild type osteoclast culture (_p_ < 0.0001). The total resorption area per osteoclast was also
significantly higher following _PolgA__mut/mut_ osteoclast culture at 2951 µm per osteoclast compared to 1737 µm of resorption on average by wild type osteoclasts (_p_ = 0.0134). High levels
of variability are observed between dentine discs within individual cell lines and also between cell lines of the same genotype for number of pits and also in the resorption area recorded
per osteoclast, particularly within the _PolgA__mut/mut_ group. RESPIRATORY CHAIN PROTEIN EXPRESSION DECLINES IN OSTEOBLASTS AND OSTEOCLASTS OF AGING MICE, AND AT AN ACCELERATED RATE IN
_POLGA_ _MUT/MUT_ MICE We have previously shown that a significant reduction in mitochondrial protein expression within osteoblasts occurs between the age of 4 and 11 months in wild type
mice, with the levels of the respiratory chain proteins COX-I and NDUFB8 levels declining by 1.8 SD and 1.9 SD respectively. In _PolgA__mut/mut_ mice aged 11 months, COX-I and NDUFB8 levels
are 13.4SD and 8.2 SD lower respectively in osteoblasts when compared to osteoblasts within wild type mice aged 4 months44. Here we investigated mitochondrial protein expression within
osteoclasts using the same methodology. Mitochondria within osteoclasts of wild type mice aged 4 months (n = 7) and 11 months (n = 7) were compared to 11 month _PolgA__mut/mut_ mice (n = 7).
The relationship between mitochondrial mass (porin), COX-I and NDUFB8 protein abundance in 4 month-old control mice is generally linear albeit with some variation, and there is evidence
that some osteoclasts express low levels of COX-I and NDUFB8 even in these young control mice. In wild type mice aged 11 months, visibly higher variation was apparent with some loss of the
linear relationship. This was even more apparent in 11 month _PolgA__mut/mut_ animals with many cells observed displaying low COX-I and NDUFB8 protein abundance (Fig. 6). In osteoclasts from
wild-type mice, mean COX-I and NDUFB8 levels decline with age by 0.76 SD and 0.36 SD respectively, from 4 to 11 months. In 11 month _PolgA__mut/mut_ osteoclasts, COX-I and NDUFB8 protein
expression in was 1.22 SD and 0.77 SD lower than in osteoclasts from wild type mice aged 4 months. When age-matched wild type and _PolgA__mut/mut_ mice were compared (both 11 months old),
COX-I and NDUFB8 levels in the latter were found to be 0.46 SD and 0.36SD lower respectively in _PolgA__mut/mut_ osteoclasts. Numerical data for individual mice is shown in Supplemental Data
S5. On average, 15.2% and 6.4% of _PolgA__mut/mut_ osteoclasts showed some degree of COX-I and NDUFB8 deficiency respectively (intermediate positive, intermediate negative or negative
classification), compared to 10.8% and 3.6% of osteoclasts from wild type mice aged 11 months. Corresponding individual graphs for all mice, depicting NDUFB8:porin and COX-I:porin ratios
following Z-score analysis, which clearly show deficiency type and severity are provided in Supplemental Data S6. Figure 7 shows representative examples of these graphs for 4 month old wild
type, 11 month old wild type and 11 month old _PolgA__mut/mut_ mice. DISCUSSION With age, somatic mtDNA mutations accumulate in mitotic and post mitotic tissue, and somatic stem cell
precursors60. We have demonstrated that osteoblasts44 osteoclast accumulate mitochondrial defects occur within ageing wild type mice and at an accelerated rate in _PolgA__mut/mut_ mice, and
that these findings correspond to increased bone loss, reduced bone formation rate, reduced osteoblast population density, and increased osteoclast population density in vivo_._ A
corresponding decline in the capacity of osteogenic cells to form mineralised matrix, and increased osteoclast resorption activity in vitro were also demonstrated. We found that exercise did
not affect bone mass or rescue the impaired function of extracted osteoblasts. Mutations and consequent dysfunction in other tissues in the _PolgA__mut/mut_ mouse model are cumulative with
age37, hence our assessment of bone phenotype at different ages. With micro CT scanning, we found a significant reduction in lumbar vertebral BV/TV in male _PolgA__mut/mut_ mice as young as
4 months and by 7 months in female _PolgA__mut/mut_ mice. With advancing age, accelerated lumbar spine bone loss continues with significant deficits shown in male _PolgA__mut/mut_ mice at 7
and 11 months, and female _PolgA__mut/mut_ mice at 11 months, compared to age and sex matched wild type mice. Femoral trabecular bone shows a similar pattern of accelerated loss in
_PolgA__mut/mut_ mice with significant reductions in BV/TV seen in females by 7 months, and males by 11 months. It is not clear why the rate of femoral trabecular bone loss appears to slow
in female _PolgA__mut/mut_ mice after the age of 7 months, with no significant difference in BV/TV observed by the age of 11 months when compared with age matched wild type mice, although
significant deficits in lumbar spine BV/TV were still apparent at this age. At each of the time points studied (4, 7 and 11 months), micro CT scan showed that BV/TV in male mice was higher
than that of females, regardless of genotype, illustrating the significant role that sex hormone composition plays in bone biology. Our data for wild type mouse femoral and vertebral BV/TV
is in keeping with previous work61 and the patterns of bone loss in _PolgA__mut/mut_ mice mirrors that seen in wild type mice, further validating the usefulness of this model. In keeping
with these findings, in vivo assessment showed significantly reduced BV/TV in _PolgA__mut/mut_ mice compared to age and sex matched wild type mice. Analysis of bone formation rates in vivo
showed significantly reduced levels of osteogenesis at the mineralisation front in _PolgA__mut/mut_ mice compared to age and sex matched wild type mice. This correlated with a significantly
reduced population density of osteoblasts observed in this mouse model. Mitochondrial dysfunction in other tissues and organs may contribute to the osteoporotic phenotype of _PolgA__mut/mut_
mice, hence our desire to study the function of osteogenic cells in vitro_,_ with the effects of whole body physiology eradicated. In keeping with the reduced bone formation rates observed
in vivo_, PolgA__mut/mut_ ALP positive osteogenic cells produced significantly less mineralised matrix in vitro than age and sex matched wild type cells. Mineralised matrix was produced in
abundance when using osteogenic cells derived from wild type bulk unfractionated skeletal mesenchymal cells in keeping with work using human and mouse MSCs62,63. However, with increasing age
of wild type mice, which accumulate mitochondrial DNA mutations at a much slower rate than their _PolgA__mut/mut_ counterparts, declining levels of mineralised matrix were produced, a
finding which correlates with increasing in vivo bone loss and reducing osteoblast mitochondrial respiratory protein expression with advancing age44. A limitation in our study is the use of
bulk unfractionated mesenchymal cells, rather than the use of purified osteogenic mesenchymal stem cells harvested using a method such as flow cytometry, and it would be useful to validate
our findings using a method such as this. MtDNA mutations are known to accumulate in stem cell populations with age64. The effect of these mtDNA mutations has been shown to vary depending on
the cell line affected65,66. There have been no previous reports of the effects on cells of the MSC lineage. Although the population densities of ALP producing cells seen in our in vitro
analysis showed no discernible difference between _PolgA__mut/mut_ and wild type cell lines, our in vivo analysis showed significant reduction in numbers of mature bone lining osteoblasts.
Clearly mitochondrial dysfunction is associated with abnormal differentiation of MSCs through an osteogenic lineage, with reduced population densities of mature bone lining osteoblasts
occurring and impaired osteogenic function, as demonstrated by reduced in vivo bone formation rates. The capacity of osteogenic cells to produce mineralised matrix in vitro is also
significantly impaired. During osteogenic differentiation of human MSCs, increasing levels of intracellular ATP content, mitochondrial DNA (mtDNA) copy number and protein subunits of
respiratory enzymes occur, which correlates with increased oxidative phosphorylation during early differentiation67,68. _Wnt_ signalling is vital for osteoblast differentiation,
proliferation and subsequent mineralisation and it also appears to be intrinsically linked to mitochondrial biogenesis69,70. This relationship appears to be mutual as when mitochondrial
biogenesis is enhanced following upregulation of TFAM activity in mouse mesenchymal cells in vitro, _Wnt_ induced β-catenin expression and osteogenesis is significantly enhanced69.
Non-weight bearing states and weightlessness in space are known to cause bone loss in humans and animal models71,72. We found no significant difference in the weight of wild type and
_PolgA__mut/mut_ mice aged 4 and 7 months to account for differences in bone loss (data not shown), and Trifunovic et. al. did not find a significant difference in body weight until the age
of 39 weeks (37). However, we acknowledge that significantly reduced body weight in _PolgA__mut/mut_ mice by the age of 11 months may contribute to accelerated bone loss at this stage.
Exercise has been shown to enhance respiratory chain activity in human muscle 42 and a similar exercise regime to ours has been shown to be protective against the ageing phenotype of
_PolgA__mut/mut_ mice when applied from the ages of 3 to 8 months, although the effects of exercise on bone loss was not evaluated40. We found that exercise did not affect bone mass in male
_PolgA__mut/mut_ mice and that osteogenic cells extracted from these mice exhibited the same reduced functional capacity as cells from non-exercised _PolgA__mut/mut_ mice of the same age.
Subtle variations between the exercise protocols used in ours and previous work may have contributed to the lack of effect observed. Recent work studying the effects of exercise on bone mass
in wild type C57BL/6 mice shows that the intensity of exercise is critical with no significant increase in bone mass occurring when intensity is too low or too high73. The authors suggest
that a minimum threshold of exercise must be reached to achieve a positive effect on bone density accrual and hypothesise that increased free radical production with excessive exercise,
impairs bone mass accumulation. Our analysis of osteoclast mitochondrial respiratory chain protein expression within osteoclasts showed that both COX-I and NDUFB8 protein expression levels
decline with advancing age in wild type mouse osteoclasts and at an accelerated rate in _PolgA__mut/mut_ mice. However, the decline was much more modest than that observed in osteoblasts 44.
These results suggest that cells of the osteoclast lineage are less vulnerable to mitochondrial dysfunction with advancing age and in the _PolgA__mut/mut_ mouse model, than cells of the
osteoblast lineage. Alternatively, osteoclasts may be more dependent on preserved mitochondrial function, perhaps meaning that only those with normal respiratory chain function develop or
survive in vivo_._ We found that the respiratory chain deficiency observed in osteoclasts is associated with an increased osteoclast population density in vivo in _PolgA__mut/mut_ mice_._
Geurts et al_._ also noted increased osteoclast numbers in vivo in this mouse model74. Increased mitochondrial biogenesis and activity has been shown to occur during osteoclast
differentiation, suggesting some degree of normal mitochondrial function is important to the process75,76,77. It is likely that a threshold of acceptable mitochondrial function exists, above
which the cell can continue to function. Stromal and osteoblast expression of RANKL and M-CSF has been shown to increase with advancing age in mice, and when these cells are used in vitro
to invoke osteoclastogenesis in osteoclast precursors, the effects are enhanced when using donor osteoblasts and stromal cells from older animals78. It is not clear whether upregulation of
these factors is related to mitochondrial dysfunction, but a potential increase of their expression in _PolgA__mut/mut_ mice, may explain the findings in our in vivo work. Similar findings
are apparent in human whole bone and cultured marrow cells, with increasing expression of RANKL and reducing expression of OPG with advancing age. Overexpression of RANKL or under expression
of OPG has been shown to lead to severe osteopenia79,80. The osteoclast progenitor pool, the expression of RANK (receptor to RANKL) and c-fms (receptor to M-CSF) have also been shown to
increase with age78,81,82. Again, it not clear whether these changes are intrinsically linked to changes in cellular mitochondrial function. Osteoclast-specific knock out of TFAM, which
results in a reduction in mitochondrial gene expression and consequent mitochondrial dysfunction, has been shown to be associated with enhanced osteoclast resorption activity despite
significantly reduced ATP content and accelerated apoptosis 38. In keeping with these findings, our in vitro work also demonstrates enhanced osteoclast activity, with significantly increased
resorption by _PolgA__mut/mut_ osteoclasts with an increased ratio of pits per osteoclast, and increased ratio of dentine resorbed per osteoclast. Variability observed between dentine discs
within individual cell lines and also between cell lines of the same genotype for number of pits and also in the resorption area recorded per osteoclast, is likely to be accounted for by
the cell culturing process, related to the harvest of variable numbers of monocytic cells when removing non-adherent cells from flasks. Whilst osteoclast differentiation and resorption is
clearly affected, the primary defect in bone homeostasis in the _PolgA__mut/mut_ mouse model, appears to be one of reduced bone formation. We believe that this is the first work to show that
mitochondrial dysfunction, a universal feature of human ageing, severely impairs osteogenesis and is associated with accelerated bone loss. Although mitochondrial dysfunction in bone cells
appears to have a direct effect on bone phenotype, the secondary effects on bone of mitochondrial dysfunction within other tissues, remains unclear. Further understanding of the potential
role of mitochondrial dysfunction in failing bone biology is crucial. Although _PolgA__mut/mut_ mice accumulate mtDNA mutations at 3–5 times of wild type mice, the resultant mitochondrial
dysfunction is sufficient by 4 months of age to cause marked dysfunction with associated bone loss. Although humans accumulate mtDNA mutations at a slower rate than this, the lifespan of
humans is such that the cumulative effects of these on bone homeostasis is a potentially significant factor in the development of osteoporosis and further investigation using human tissues
and cell lines is warranted. REFERENCES * Rodan, G. A. & Martin, T. J. Therapeutic approaches to bone diseases. _Science_ 289, 1508–1514 (2000). ADS CAS PubMed Google Scholar *
Karsenty, G. & Wagner, E. F. Reaching a genetic and molecular understanding of skeletal development. _Dev. Cell_ 2, 389–406 (2002). CAS PubMed Google Scholar * Hendrickx, G., Boudin,
E. & Van Hul, W. A look behind the scenes: the risk and pathogenesis of primary osteoporosis. _Nat. Rev. Rheumatol._ 11, 462–474 (2015). PubMed Google Scholar * Matkovic, V. _et al._
Timing of peak bone mass in Caucasian females and its implication for the prevention of osteoporosis. Inference from a cross-sectional model. _J. Clin. Invest._ 93, 799–808 (1994). CAS
PubMed PubMed Central Google Scholar * Shanbhogue, V. V., Brixen, K. & Hansen, S. Age- and sex-related changes in bone microarchitecture and estimated strength: a three-year
prospective study using HRpQCT. _J. Bone Miner. Res._ 31, 1541–1549 (2016). PubMed Google Scholar * Hung, V. W. Y. _et al._ Age-related differences in volumetric bone mineral density,
microarchitecture, and bone strength of distal radius and tibia in Chinese women: a high-resolution pQCT reference database study. _Osteoporos. Int._ 26, 1691–1703 (2015). CAS PubMed
Google Scholar * Amin, S. & Khosla, S. Sex- and age-related differences in bone microarchitecture in men relative to women assessed by high-resolution peripheral quantitative computed
tomography. _J. Osteoporos._ 2012, 129760–129760 (2012). PubMed PubMed Central Google Scholar * Felson, D. T. _et al._ The effect of postmenopausal estrogen therapy on bone density in
elderly women. _N. Engl. J. Med._ 329, 1141–1146 (1993). CAS PubMed Google Scholar * Kanis, J. A. Diagnosis of osteoporosis and assessment of fracture risk. _Lancet_ 359, 1929–1936
(2002). PubMed Google Scholar * Mayhew, P. M. _et al._ Relation between age, femoral neck cortical stability, and hip fracture risk. _Lancet_ 366, 129–135 (2005). PubMed Google Scholar *
O’Brien, C. A. _et al._ Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. _Endocrinology_ 145, 1835–1841 (2004).
PubMed Google Scholar * Chiodini, I. _et al._ Subclinical hypercortisolism among outpatients referred for osteoporosis. _Ann. Intern. Med._ 147, 541–548 (2007). PubMed Google Scholar *
Fink, H. A. _et al._ Association of testosterone and estradiol deficiency with osteoporosis and rapid bone loss in older men. _J. Clin. Endocrinol. Metab._ 91, 3908–3915 (2006). CAS PubMed
Google Scholar * Baran, D. T., Bergfeld, M. A., Teitelbaum, S. L. & Avioli, L. V. Effect of testosterone therapy on bone formation in an osteoporotic hypogonadal male. _Calcif. Tissue
Res._ 26, 103–106 (1978). CAS PubMed Google Scholar * Aitken, J. M., Armstrong, E. & Anderson, J. B. Osteoporosis after oophorectomy in the mature female rat and the effect of
oestrogen and/or progestogen replacement therapy in its prevention. _J. Endocrinol._ 55, 79–87 (1972). CAS PubMed Google Scholar * Lindsay, R. _et al._ Long-term prevention of
postmenopausal osteoporosis by oestrogen. _Lancet_ 307, 1038–1041 (1976). Google Scholar * Prince, R. L. _et al._ Prevention of postmenopausal osteoporosis: a comparative study of exercise,
calcium supplementation, and hormone-replacement therapy. _N. Engl. J. Med._ 325, 1189–1195 (1991). CAS PubMed Google Scholar * Heinonen, A. _et al._ Randomised controlled trial of
effect of high-impact exercise on selected risk factors for osteoporotic fractures. _Lancet_ 348, 1343–1347 (1996). CAS PubMed Google Scholar * Karlsson, M. K. _et al._ Exercise during
growth and bone mineral density and fractures in old age. _Lancet_ 355, 469–470 (2000). CAS PubMed Google Scholar * Kurland, E. S. _et al._ Insulin-like growth factor-I in men with
idiopathic osteoporosis 1. _J. Clin. Endocrinol. Metab._ 82, 2799–2805 (1997). CAS PubMed Google Scholar * Ljunghall, S. _et al._ Low plasma levels of insulin-like growth factor 1 (IGF-1)
in male patients with idiopathic osteoporosis. _J. Intern. Med._ 232, 59–64 (1992). CAS PubMed Google Scholar * Cortopassi, G. A. & Arnheim, N. Detection of a specific mitochondrial
DNA deletion in tissues of older humans. _Nucl. Acids Res._ 18, 6927–6933 (1990). CAS PubMed PubMed Central Google Scholar * Cortopassi, G. A., Shibata, D., Soong, N. W. & Arnheim,
N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. _Proc. Natl. Acad. Sci. USA_ 89, 7370–7374 (1992). ADS CAS PubMed PubMed Central Google
Scholar * Muller-Hocker, J. Cytochrome-c-oxidase deficient cardiomyocytes in the human heart—an age-related phenomenon. A histochemical ultracytochemical study. _Am. J. Pathol._ 134,
1167–1173 (1989). CAS PubMed PubMed Central Google Scholar * Cottrell, D. A. _et al._ Cytochrome c oxidase deficient cells accumulate in the hippocampus and choroid plexus with age.
_Neurobiol. Aging_ 22, 265–272 (2001). CAS PubMed Google Scholar * Taylor, R. W. _et al._ Mitochondrial DNA mutations in human colonic crypt stem cells. _J. Clin. Invest._ 112, 1351–1360
(2003). CAS PubMed PubMed Central Google Scholar * Fellous, T. G. _et al._ Locating the stem cell niche and tracing hepatocyte lineages in human liver. _Hepatology_ 49, 1655–1663 (2009).
CAS PubMed Google Scholar * Kujoth, G. C. _et al._ Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. _Science_ 309, 481–484 (2005). ADS CAS PubMed
Google Scholar * Linnane, A., Ozawa, T., Marzuki, S. & Tanaka, M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. _Lancet_ 333, 642–645
(1989). Google Scholar * Larsson, N.-G. Somatic mitochondrial DNA mutations in mammalian aging. _Annu. Rev. Biochem._ 79, 683–706 (2010). CAS PubMed Google Scholar * Senior, A. E. ATP
synthesis by oxidative phosphorylation. _Physiol. Rev._ 68, 177–231 (1988). CAS PubMed Google Scholar * Lunt, S. Y. & Vander Heiden, M. G. Aerobic glycolysis: meeting the metabolic
requirements of cell proliferation. _Annu. Rev. Cell Dev. Biol._ 27, 441–464 (2011). CAS PubMed Google Scholar * Hayashi, J. _et al._ Introduction of disease-related mitochondrial DNA
deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. _Proc. Natl. Acad. Sci. USA_ 88, 10614–10618 (1991). ADS CAS PubMed PubMed Central Google
Scholar * Chomyn, A. _et al._ MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of
upstream and downstream mature transcripts. _Proc. Natl. Acad. Sci. USA_ 89, 4221–4225 (1992). ADS CAS PubMed PubMed Central Google Scholar * Hance, N., Ekstrand, M. I. &
Trifunovic, A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. _Hum. Mol. Genet._ 14, 1775–1783 (2005). CAS PubMed Google Scholar * Kaguni, L. S. DNA
Polymerase γ, The mitochondrial replicase. _Annu. Rev. Biochem._ 73, 293–320 (2004). CAS PubMed Google Scholar * Trifunovic, A. _et al._ Premature ageing in mice expressing defective
mitochondrial DNA polymerase. _Nature_ 429, 417–423 (2004). ADS CAS PubMed Google Scholar * Miyazaki, T. _et al._ Intracellular and extracellular ATP coordinately regulate the inverse
correlation between osteoclast survival and bone resorption. _J. Biol. Chem._ 287, 37808–37823 (2012). CAS PubMed PubMed Central Google Scholar * Kobayashi, K. _et al._ Mitochondrial
superoxide in osteocytes perturbs canalicular networks in the setting of age-related osteoporosis. _Sci. Rep._ 5, 9148 (2015). CAS PubMed PubMed Central Google Scholar * Safdar, A. _et
al._ Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. _Proc. Natl. Acad. Sci. USA_ 108, 4135–4140 (2011). ADS CAS PubMed
PubMed Central Google Scholar * Morgan, T. E., Cobb, L. A., Short, F. A., Ross, R. & Gunn, D. R. In _Muscle Metabolism During Exercise: Proceedings of a Karolinska Institutet Symposium
held in Stockholm, Sweden, September 6–9, 1970_ (eds Bengt Pernow & Bengt Saltin) 87–95 (Springer US, 1971). * Menshikova, E. V. _et al._ Effects of exercise on mitochondrial content
and function in aging human skeletal muscle. _J. Gerontol. Ser. A Biol. Sci. Med. Sci._ 61, 534–540 (2006). Google Scholar * Harada, S. & Rodan, G. A. Control of osteoblast function and
regulation of bone mass. _Nature_ 423, 349–355 (2003). ADS CAS PubMed Google Scholar * Dobson, P. F. _et al._ Unique quadruple immunofluorescence assay demonstrates mitochondrial
respiratory chain dysfunction in osteoblasts of aged and PolgA−/− mice. _Sci. Rep._ 6, 31907 (2016). ADS CAS PubMed PubMed Central Google Scholar * Ross, J. M. _et al._ Germline
mitochondrial DNA mutations aggravate ageing and can impair brain development. _Nature_ 501, 412–415 (2013). ADS CAS PubMed PubMed Central Google Scholar * Dennis, E. P. _et al._ CRELD2
is a novel LRP1 chaperone that regulates noncanonical WNT signaling in skeletal development. _J. Bone Miner. Res._ (2020). https://doi.org/10.1002/jbmr.4010. Article PubMed Google Scholar
* Orriss, I. R., Hajjawi, M. O., Huesa, C., MacRae, V. E. & Arnett, T. R. Optimisation of the differing conditions required for bone formation in vitro by primary osteoblasts from mice
and rats. _Int. J. Mol. Med._ 34, 1201–1208 (2014). CAS PubMed PubMed Central Google Scholar * Kawamura, N. _et al._ Akt1 in osteoblasts and osteoclasts controls bone remodeling. _PLoS
ONE_ 2, e1058–e1058 (2007). ADS PubMed PubMed Central Google Scholar * Wang, J. _et al._ miR-30e reciprocally regulates the differentiation of adipocytes and osteoblasts by directly
targeting low-density lipoprotein receptor-related protein 6. _Cell Death Dis._ 4, e845 (2013). CAS PubMed PubMed Central Google Scholar * Eskildsen, T. _et al._ MicroRNA-138 regulates
osteogenic differentiation of human stromal (mesenchymal) stem cells in vivo. _Proc. Natl. Acad. Sci._ 108, 6139 (2011). ADS PubMed PubMed Central Google Scholar * Smink, J. J. _et al._
Transcription factor C/EBPbeta isoform ratio regulates osteoclastogenesis through MafB. _EMBO J._ 28, 1769–1781 (2009). CAS PubMed PubMed Central Google Scholar * Taylor, S. E. B., Shah,
M. & Orriss, I. R. Generation of rodent and human osteoblasts. _BoneKEy Rep._ 3, 585–585 (2014). PubMed PubMed Central Google Scholar * Liu, W. _et al._ GDF11 decreases bone mass by
stimulating osteoclastogenesis and inhibiting osteoblast differentiation. _Nat. Commun._ 7, 12794–12794 (2016). ADS CAS PubMed PubMed Central Google Scholar * Murshed, M., Harmey, D.,
Millán, J. L., McKee, M. D. & Karsenty, G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. _Genes Dev._
19, 1093–1104 (2005). CAS PubMed PubMed Central Google Scholar * Wennberg, C. _et al._ Functional characterization of osteoblasts and osteoclasts from alkaline phosphatase knockout mice.
_J. Bone Miner. Res._ 15, 1879–1888 (2000). CAS PubMed Google Scholar * Orriss, I. R. & Arnett, T. R. Rodent osteoclast cultures. _Methods Mol. Biol._ 816, 103–117 (2012). CAS
PubMed Google Scholar * Blachly-Dyson, E., Baldini, A., Litt, M., McCabe, E. R. & Forte, M. Human genes encoding the voltage-dependent anion channel (VDAC) of the outer mitochondrial
membrane: mapping and identification of two new isoforms. _Genomics_ 20, 62–67 (1994). CAS PubMed Google Scholar * Hirst, J., Carroll, J., Fearnley, I. M., Shannon, R. J. & Walker, J.
E. The nuclear encoded subunits of complex I from bovine heart mitochondria. _Biochem. Biophys. Acta_ 1604, 135–150 (2003). CAS PubMed Google Scholar * Tsukihara, T. _et al._ The whole
structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. _Science_ 272, 1136–1144 (1996). ADS CAS PubMed Google Scholar * Taylor, R. W. & Turnbull, D. M. Mitochondrial DNA
mutations in human disease. _Nat. Rev. Genet._ 6, 389–402 (2005). CAS PubMed PubMed Central Google Scholar * Glatt, V., Canalis, E., Stadmeyer, L. & Bouxsein, M. L. Age-related
changes in trabecular architecture differ in female and male C57BL/6J mice. _J. Bone Miner. Res._ 22, 1197–1207 (2007). PubMed Google Scholar * Bergman, R. J. _et al._ Age-related changes
in osteogenic stem cells in mice. _J. Bone Miner. Res._ 11, 568–577 (1996). CAS PubMed Google Scholar * Stenderup, K., Justesen, J., Clausen, C. & Kassem, M. Aging is associated with
decreased maximal life span and accelerated senescence of bone marrow stromal cells. _Bone_ 33, 919–926 (2003). PubMed Google Scholar * Baines, H. L., Turnbull, D. M. & Greaves, L. C.
Human stem cell aging: do mitochondrial DNA mutations have a causal role?. _Aging Cell_ 13, 201–205 (2014). CAS PubMed PubMed Central Google Scholar * Norddahl, G. L. _et al._
Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. _Cell Stem Cell_ 8, 499–510 (2011). CAS PubMed Google
Scholar * Ahlqvist, K. J. _et al._ Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in polg mutator mice. _Cell Metab._ 15, 100–109
(2012). CAS PubMed Google Scholar * Pietilä, M. _et al._ Mitochondrial function determines the viability and osteogenic potency of human mesenchymal stem cells. _Tissue Eng. Part C
Methods_ 16, 435–445 (2010). PubMed Google Scholar * Chen, C. T., Shih, Y. R., Kuo, T. K., Lee, O. K. & Wei, Y. H. Coordinated changes of mitochondrial biogenesis and antioxidant
enzymes during osteogenic differentiation of human mesenchymal stem cells. _Stem Cells_ 26, 960–968 (2008). CAS PubMed Google Scholar * An, J. H. _et al._ Enhanced mitochondrial
biogenesis contributes to Wnt induced osteoblastic differentiation of C3H10T1/2 cells. _Bone_ 47, 140–150 (2010). CAS PubMed Google Scholar * Scarpulla, R. C. Transcriptional paradigms in
mammalian mitochondrial biogenesis and function. _Physiol. Rev._ 88, 611–638 (2008). CAS PubMed Google Scholar * Taaffe, D. R. _et al._ Differential effects of swimming versus
weight-bearing activity on bone mineral status of eumenorrheic athletes. _J. Bone Miner. Res._ 10, 586–593 (1995). CAS PubMed Google Scholar * Vico, L. _et al._ Effects of long-term
microgravity exposure on cancellous and cortical weight-bearing bones of cosmonauts. _Lancet_ 355, 1607–1611 (2000). CAS PubMed Google Scholar * Zhang, L. _et al._ The effects of
different intensities of exercise and active vitamin D on mouse bone mass and bone strength. _J. Bone Miner. Metab._ 35, 265–277 (2017). CAS PubMed Google Scholar * Geurts, J. _et al._
Prematurely aging mitochondrial DNA mutator mice display subchondral osteopenia and chondrocyte hypertrophy without further osteoarthritis features. _Sci. Rep._ 10, 1296 (2020). ADS CAS
PubMed PubMed Central Google Scholar * Lemma, S. _et al._ Energy metabolism in osteoclast formation and activity. _Int. J. Biochem. Cell Biol._ 79, 168–180 (2016). CAS PubMed Google
Scholar * Czupalla, C., Mansukoski, H., Pursche, T., Krause, E. & Hoflack, B. Comparative study of protein and mRNA expression during osteoclastogenesis. _Proteomics_ 5, 3868–3875
(2005). CAS PubMed Google Scholar * Ishii, K. A. _et al._ Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. _Nat. Med._ 15, 259–266 (2009).
CAS PubMed Google Scholar * Cao, J. J. _et al._ Aging increases stromal/osteoblastic cell-induced osteoclastogenesis and alters the osteoclast precursor pool in the mouse. _J. Bone Miner.
Res._ 20, 1659–1668 (2005). CAS PubMed Google Scholar * Fazzalari, N. L., Kuliwaba, J. S., Atkins, G. J., Forwood, M. R. & Findlay, D. M. The ratio of messenger RNA levels of
receptor activator of nuclear factor kappaB ligand to osteoprotegerin correlates with bone remodeling indices in normal human cancellous bone but not in osteoarthritis. _J. Bone Miner. Res._
16, 1015–1027 (2001). CAS PubMed Google Scholar * Makhluf, H. A., Mueller, S. M., Mizuno, S. & Glowacki, J. Age-related decline in osteoprotegerin expression by human bone marrow
cells cultured in three-dimensional collagen sponges. _Biochem. Biophys. Res. Commun._ 268, 669–672 (2000). CAS PubMed Google Scholar * Perkins, S. L., Gibbons, R., Kling, S. & Kahn,
A. J. Age-related bone loss in mice is associated with an increased osteoclast progenitor pool. _Bone_ 15, 65–72 (1994). CAS PubMed Google Scholar * Koshihara, Y. _et al._
Osteoclastogenic potential of bone marrow cells increases with age in elderly women with fracture. _Mech. Ageing Dev._ 123, 1321–1331 (2002). PubMed Google Scholar Download references
ACKNOWLEDGEMENTS This work was supported by The Wellcome Centre for Mitochondrial Research [G906919], Newcastle University Centre for Ageing and Vitality (supported by the Biotechnology and
Biological Sciences Research Council and MRC [G016354/1]), the Malhotra Group, Newcastle NIHR Biomedical Research Centre in Age and Age Related Diseases award to the Newcastle upon Tyne
Hospitals NHS Foundation Trust. The Royal College of Surgeons, England provided a one year Fellowship to support this work (RS226). Bone scans were performed by Professor Rob Van T’Hof,
Gemma Charlesworth and Amanda Prior at Institute of Ageing and Chronic Disease, Liverpool University, UK. AUTHOR INFORMATION Author notes * These authors contributed equally: David J.
Deehan, Doug M. Turnbull and Laura C. Greaves. AUTHORS AND AFFILIATIONS * Wellcome Centre for Mitochondrial Research, Newcastle University, Newcastle upon Tyne, NE2 4HH, UK Philip F. Dobson,
Daniel Hipps, Amy Reeve, Carla Bradshaw, Craig Stamp, Anna Smith, Doug M. Turnbull & Laura C. Greaves * Translational and Clinical Research Institute, Newcastle University, Newcastle
upon Tyne, NE2 4HH, UK Philip F. Dobson, Daniel Hipps, Amy Reeve, David J. Deehan & Doug M. Turnbull * Biosciences Institute, Newcastle University, Newcastle upon Tyne, NE2 4HH, UK Ella
P. Dennis, Carla Bradshaw, Craig Stamp, Anna Smith & Laura C. Greaves * Bioimaging Unit, Medical School, FMS Professional Services, Newcastle University, Newcastle upon Tyne, NE2 4HH, UK
Alex Laude Authors * Philip F. Dobson View author publications You can also search for this author inPubMed Google Scholar * Ella P. Dennis View author publications You can also search for
this author inPubMed Google Scholar * Daniel Hipps View author publications You can also search for this author inPubMed Google Scholar * Amy Reeve View author publications You can also
search for this author inPubMed Google Scholar * Alex Laude View author publications You can also search for this author inPubMed Google Scholar * Carla Bradshaw View author publications You
can also search for this author inPubMed Google Scholar * Craig Stamp View author publications You can also search for this author inPubMed Google Scholar * Anna Smith View author
publications You can also search for this author inPubMed Google Scholar * David J. Deehan View author publications You can also search for this author inPubMed Google Scholar * Doug M.
Turnbull View author publications You can also search for this author inPubMed Google Scholar * Laura C. Greaves View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS P.F.D. conducted experimental work and data analysis and wrote the manuscript. E.P.D. conducted experimental work and data analysis. A.R., A.L. and D.H. provided
assistance with image analysis. L.C.G., C.S. and C.B. bred, exercised and genotyped the mouse colony. C.B. performed in vivo procedures. A.S provided assistance with tissue sectioning.
L.C.G., D.J.D. and D.M.T. designed and supervised the study and revised the manuscript. All authors approved the final submitted version. CORRESPONDING AUTHORS Correspondence to Doug M.
Turnbull or Laura C. Greaves. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE Springer Nature remains neutral
with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION 1. RIGHTS AND PERMISSIONS OPEN ACCESS This article
is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you
give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material
in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Dobson, P.F., Dennis, E.P., Hipps, D. _et al._
Mitochondrial dysfunction impairs osteogenesis, increases osteoclast activity, and accelerates age related bone loss. _Sci Rep_ 10, 11643 (2020). https://doi.org/10.1038/s41598-020-68566-2
Download citation * Received: 07 March 2019 * Accepted: 24 June 2020 * Published: 15 July 2020 * DOI: https://doi.org/10.1038/s41598-020-68566-2 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative