Transcriptome profiling of human oocytes experiencing recurrent total fertilization failure
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
There exist some patients who face recurrent total fertilization failure during assisted reproduction treatment, but the pathological mechanism underlying is elusive. Here, by using
sc-RNA-seq method, the transcriptome profiles of ten abnormally fertilized zygotes were assessed, including five zygotes from one patient with recurrent Poly-PN zygotes, and five zygotes
from a patient with pronuclear fusion failure. Four zygotes with three pronuclear (Tri-PN) were collected from four different patients as controls. After that, we identified 951 and 1697
significantly differentially expressed genes (SDEGs) in Poly-PN and PN arrest zygotes, respectively as compared with the control group. KEGG analyses indicated down regulated genes in the
Poly-PN group included oocyte meiosis related genes, such as PPP2R1B, YWHAZ, MAD2L1, SPDYC, SKP1 and CDC27, together with genes associated with RNA processing, such as SF3B1, LOC645691,
MAGOHB, PHF5A, PRPF18, DDX5, THOC1 and BAT1. In contrast, down regulated genes in the PN arrest group, included cell cycle genes, such as E2F4, DBF4, YWHAB, SKP2, CDC23, SMC3, CDC25A, CCND3,
BUB1B, MDM2, CCNA2 and CDC7, together with homologous recombination related genes, such as NBN, XRCC3, SHFM1, RAD54B and RAD51. Thus, our work provides a better understanding of
transcriptome profiles underlying RTFF, although it based on a limited number of patients.
Despite nearly forty years of scientific and clinical advance in the field of assisted reproduction, there still exist some rarely patients, even though rarely occur, who have to face
recurrent total fertilization failure (RTFF) without any visual precautionary indicator1,2,3, even some of them could be rescued by assisted oocyte activation4. Therefore, elucidating the
internal mechanism of fertilization failure is of great importance for these patients. However, until now, the relevant etiological analysis was often restricted to morphology during IVF,
such as immature oocytes5, improper meiosis6, zygotes with abnormal pronuclei7, and di-pronuclear zygote failing mitotic cleavage8. Due to small amount of material available, deciphering
mechanisms underlying these defects remain technical challenging.
Recent technical advances in single-cell sequencing open a new era for exploring the biological state of a single cell at both the DNA and RNA levels for studying variations in genome9,10,
transcriptome, and epigenome11 separately or in parallel12. Originally adopted by Surani’s research team13, this approach has been applied successfully in discriminating cell
types14,15,16,17,18,19, elucidating regulatory circuits20, and investigating tumor heterogeneity21,22. In reproductive biology fields, this technique has been used for screening
transcriptome of tissues23 and germline cells at different stages24,25,26,27,28,29. The single cell sequencing technique has great potential in clinical implication30,31,32, especially in
the diagnosis for clarifying the molecular mechanisms of fertilization failure at a single cell resolution. So the aim of this work was to characterize the pathological changes of human
zygotes with RTFF at the transcriptional level.
As clinical treatment history shown (Table 1), one patient experienced two stimulated cycles under different procedures with 4 and 5 Poly-PN fertilized eggs after ICSI treatment,
respectively. Another patient had all zygotes with PN arrest, with 18, 7 and 9 matured oocytes retrieved separately in three cycles although three different ovarian stimulation procedures
were employed each time. Moreover, There were no significant differences in serum levels of FSH, LH, E2 and progesterone at baseline and trigger day in patients with Poly-PN, PN arrest, and
the control groups (Supplementary Fig. 1), indicating that the observed defects in the zygotes were more likely associated with oocyte original molecular defects rather than ovarian
stimulation protocol.
It is crucial for oocyte to accumulate indispensable mRNAs to ensure its later use for fertilization and subsequent cell division before the zygotic gene activation33. As the scarce of the
oocytes for RTFF patients, it was difficult to collect enough donated oocytes for our study. Therefore, we investigate the transcriptome profile using the unfertilized oocyte after clinical
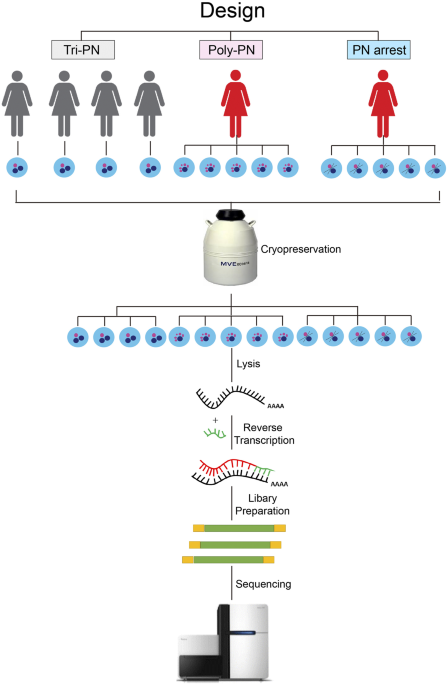
treatment. The procedure of our work was shown in Fig. 1. After sequencing using Illumina HiSeq 2,500 sequencer, we obtain about 142 million clean reads, of which 116 million clean reads
mapped to human genome reference. On average, 15,058, 14,995 and 17,713 genes (FPKM ≥ 0.1), 10,471, 10,451 and 10,289 genes (FPKM ≥ 1) or 4,539, 4,410 and 3,630 genes (FPKM ≥ 10) were
acquired in Tri-PN, Poly-PN and PN arrest groups, respectively (Fig. 2a). These results were consistent with the data from a previous study24, implying that our technology has the similar
sensitivity and coverage.
Transcriptome profile of the Poly-PN and PN-arrest zygotes. (a) The number of reference transcripts averagely found in each sample in different groups based on clustering of expression
patterns for 14 single cell samples from Poly-PN, PN arrest and Control groups; (c,d) Scatterplot showing the number of genes up regulated (red) and down regulated (blue) in Poly PN (c) and
PN arrest zygotes (d) separately compared with control group.
To compare the global transcriptome profiles of unfertilized eggs or zygotes among different groups, we analyzed data by hierarchical clustering, and the results indicated that 14 zygotes
from 3 groups were clustered into corresponding groups and separated from each other (Fig. 2b). Four Tri-PN zygotes from different patients have the similar transcriptome profile in spite of
the heterogeneity in patient source. Interestingly, we also found that five PN arrest zygotes clustered closer with four Tri-PN zygotes, but away from the five Poly-PN zygotes. This finding
indicated that the underlying mechanisms was quite different between the Poly-PN and PN arrest group.
To clarify underlying mechanisms, we analyzed major differences of expression profile among different groups. To rule out technical errors causing artifacts of gene expression, all reference
genes with average FPKM > 0.5 in any of three groups were used for subsequent analysis. According to the criteria of fold change > 2 or