Distortion-stabilized ordered structures in A2BB’O7 mixed pyrochlores
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Pyrochlore oxides (A2B2O7) are interesting for a number of technological applications, including radiation damage tolerance and as ionic conductors. Mixed pyrochlores—containing two A and/or
two B site cations—provide even more flexibility for tailoring properties owing to the diverse chemical and configurational degrees of freedom accessible within this chemical space. Here,
we examine relative stability of different cation orderings in one model double pyrochlore Gd2(ZrxTi1−x)2O7, as a function of Zr content x. Our results show that, in the presence of some
very specific local cation arrangements, certain cation-ordered compositions in this system are highly stabilized as a result of large oxygen relaxation displacements, leading to the
formation of an ordered ‘double’ pyrochlore structure. The origins of these anomalous oxygen relaxations are traced back to both the local cation symmetry and a strong chemical preference of
Zr atoms towards adopting a 7-fold coordination environment, as opposed to a 6-fold coordination available in a regular pyrochlore structure. Subsequently, we examine the stability of this
type of ordering in 131 other pyrochlore compositions. Implications of our findings are discussed in relation to the observed composition-dependent ionic conductivity in these systems and
connections with previously reported experimental findings are made.
Oxides are an attractive class of materials due to their extreme variety in functionality. This richness arises from the very flexible chemistry of these materials. Indeed, beyond base
compounds such as binaries and ternaries, the possibilities for modifying the chemistry of most oxides is nearly limitless. Such multicomponent oxides often have significantly improved
performance over the end-member compounds, with enhanced scintillation,1 ferroelectricity,2,3 piezoelectricity,4 high temperature stability5 and catalytic response.6 By expanding the
chemical space in which compound discovery can occur by considering mixed oxides, the potential for materials discovery expands exponentially.
Thus, in the quest for new materials, understanding the structures that result from mixing simpler compounds to form more complex materials is critical. This has been extensively studied in
some cases. For example, in the perovskite family of compounds (ABO3), the properties of so-called double perovskites have been examined by multiple groups.7,8 These compounds, containing
two A and/or two B cations, can form ordered structures for some chemistries9,10,11 and these orderings are known to modify functional properties, including oxygen transport12,13 and
ferromagnetism.14
In the case of pyrochlores (A2B2O7), the subject of the current work, mixed systems have been used to understand, amongst other properties, the role of disorder in both radiation damage
tolerance and ionic conductivity. Simply, as, for example, the B cation is gradually changed from Ti and Zr, the propensity for cation disorder increases and this has been correlated to the
functionality of the material. In materials such as Gd2(ZrxTi1−x)2O7 (GZTO), as the chemistry shifts from x = 0 to x = 1, disorder increases and so does the resistance to
amorphization15,16,17 and the magnitude of ionic conductivity.18 Thus, in many studies of ionic conductivity, disorder is induced in the system via the introduction of additional
cations.18,19,20 These studies have led to conflicting results, with some showing a direct increase in the conductivity vs. disorder18 while others suggest that the ordered phase has a
higher conductivity than the disordered state.19,20
Implicitly, these studies assume a smooth and gradual transition from the ordered state to the disordered state with changes in chemistry. However, this is not always the case. For example,
NMR studies show that, depending on both the A and B chemistry of the compound, mixed pyrochlores can form either solid solutions or phase separate.21,22 This complicates the establishment
of structure-property relationships with disorder.
In this work, we consider a further complication, the possibility that, even in cases where the cations form a solid solution in mixed pyrochlores, they may form ordered structures,
reminiscent of the double perovskites, rather than a disordered solid solution. These ordered structures would naturally exhibit different transport behavior than a random solid solution, as
we have seen previously for double perovskites.13 Thus, in any campaign to establish structure-property relationships for mixed pyrochlores, one must have a solid understanding of what the
detailed atomic structure of the mixed pyrochlore is. We use density functional theory (DFT) to investigate the possible orderings as a function of x in Gd2(ZrxTi1−x)2O7. We identify unique
ordered ground states that are stabilized by significant distortions in the oxygen sublattice, distortions that increase the coordination of Zr from 6 to 7. We then examine the stability of
this ordering in other pyrochlore compositions. We find that in many compositions in which the A cation is of sufficient size and the B sublattice contains sufficient Zr and/or Hf, ordered
structures are thermodynamically preferred, at least at low temperature. These results offer new insight into the structures that drive performance in mixed pyrochlores.
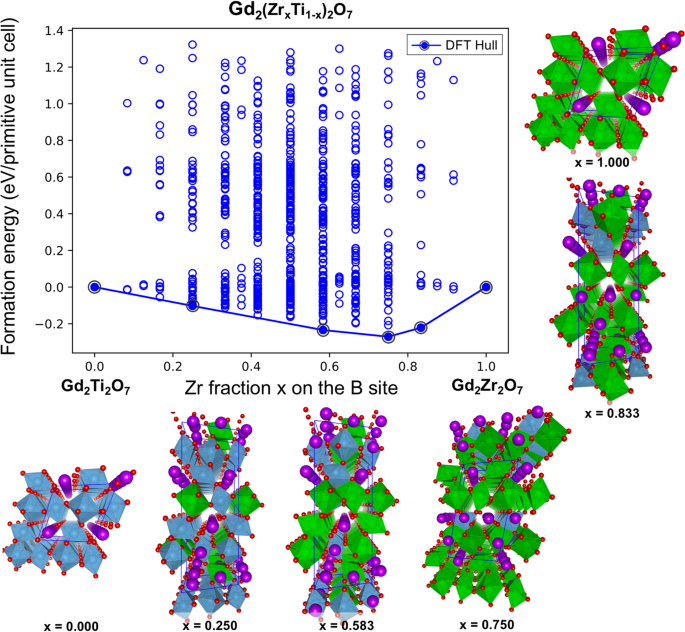
The DFT-computed mixing energies (i.e., the formation energies with respect to the pure pyrochlore chemistries with x = 0 and x = 1) for the entire set of 704 configurations enumerated
within the generalized Gd2(ZrxTi1−x)2O7 formula unit are presented in Fig. 1. In addition to the end points, the DFT-computed convex hull identifies four different ground-state structures
with compositions x = 0.25, 0.583, 0.75 and 0.833 (the relaxed geometries are provided in the Supporting Information). The atomistic structures for the identified ground-state ordered
structures are also depicted in Fig. 1. The DFT results in Fig. 1 indicate that, as the composition varies from Ti rich to Zr rich, there are ordered structures of Ti and Zr on the
B-sublattice that are thermodynamically stable. Thus, similar to double perovskites, these mixed pyrochlore systems may also exhibit some tendency to form ordered solid solutions. Further,
the propensity for ordering is greatest for a Zr-rich composition of x = 0.75, indicating that the propensity for forming an ordered structure is sensitive to the composition of the mixed
system.
Calculated mixing energies (i.e., formation energies relative to the pure single pyrochlore end point compositions with x = 0 and x = 1) per primitive unit cell (with 22 atoms, containing
two effective pyrochlore formula units) for the entire set of 704 unique configurations enumerated within the Gd2(ZrxTi1−x)2O7 chemistries. The DFT predicted convex hull is represented as a
solid line. The atomistic structures for the identified ground-state ordered structures on the convex hull are illustrated. While Gd and O atoms are represented by purple and red spheres, Ti
and Zr polyhedra are depicted in blue and green colors, respectively
We further note that the specific orderings identified in Fig. 1 between x = 0 and x = 1 (i.e., mixed pyrochlores) all have a superstructure that is longer-ranged than simple “single”
pyrochlore structure. In other words, the orderings are not possible with the standard pyrochlore primitive unit cell containing 22 atoms (i.e., two formula units) and need either 44-atom
(for x = 0.75) or 66-atom supercells (for x = 0.25, 0.583, and 0.833). To look for specific signatures for the aforementioned cation ordering, in Fig. 2 we compare the X-ray diffraction
(XRD) patterns for mixed GZTO (using the DFT-computed ground-state structure with x = 0.75 that exhibits the lowest mixing energy) and GZO pyrochlores. It can be seen that as a consequence
of B-site ordering some of the peaks in the “single” pyrochlore GZO are split in the mixed GZTO structure. Furthermore, we also find some new (weak) peaks at various angles that are a
reflection of the ordering in the mixed structure.
The DFT calculations provide the zero-Kelvin ground-state structure and a range of higher-energy metastable structures for each composition. Thus, while the ground states are ordered
structures, it is possible that their range of stability versus temperature is rather limited. One approach to understand the temperature-dependent phase stability of these structures is to
use Monte Carlo. Given the expense of doing Monte Carlo directly with DFT energetics, we turn to CE to parameterize an effective Hamiltonian.
As a next step, we use the DFT computations to fit a CE Hamiltonian to describe energetics of any given atomic configuration and composition x for the B-site ordered pyrochlore chemistries.
We included the entire set of 704 configurations in the CE fitting while closely following the approach previously described.23 During the effective cluster interactions (ECI) optimization
procedure, we systematically considered clusters with up to n-body interactions with n ∈ [4, 7] while gradually varying the maximum allowed site spacing between the n-body clusters from 7 Å
to 10 Å. Our CE analysis showed that while CE fits with small and moderate basis sets failed to describe the DFT grounds states, ECIs chosen from a much larger set (with up to 7-body
interactions and maximum allowed site spacing of 10 Å) were able to adequately capture the relative energetics of the structures in the CE training set (see Fig. S1 in the Supporting
Information). Although the adopted ECI optimization procedure explicitly included a k-fold cross-validation strategy to select the best set of ECIs (in order to avoid overfitting to the
training data and thereby leading to a better generalizability on unseen configurations), we found that the unusually large basis sets that were eventually able to provide a good fit for the
entire set of low-energy DFT configurations inevitably led to an overfitting of the training data.
To further understand the poor performance of the CE fits to the DFT-computed energetics, we looked into basis and lattice relaxation patterns of the DFT configurations used in the fitting
procedure. Our analysis identifies large basis deformations for a subset of the compounds as a cause of this problem. More specifically, we find that in certain local chemical environments O
atoms display unusually large relaxation displacements (~1.5 Å), which not only lower the energies and volumes of the supercells significantly, but also result in local coordination
environment changes of the nearby cations. Essentially, a subset of oxygen atoms displace away from their initial pyrochlore lattice positions, forming a structure that does not correspond
exactly to that of pyrochlore. Such relaxations have not been previously reported for any pyrochlore chemistries, to the best of our knowledge.
In order to quantify the extent of the basis deformations resulting from the anomalous O relaxations, we define a cost function that describes the degree to which basis sites have relaxed
with respect to the ideal pyrochlore structure. The basis deformation cost function is defined as the mean squared displacement of atoms from their position in the relaxed ideal structure
(i.e., a reference structure accounting for the lattice relaxations but the internal coordinates fixed to the unrelaxed ideal positions). Figure 3a, b present formation energies and volumes
of the relaxed structures per 22 atom primitive cell as a function of composition, with the marker colors representing the extent of the basis deformation. It is interesting to note that
while ground-state compounds with x