Inhibition of GPX4 enhances CDK4/6 inhibitor and endocrine therapy activity in breast cancer
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
CDK4/6 inhibition in combination with endocrine therapy is the standard of care for estrogen receptor (ER+) breast cancer, and although cytostasis is frequently observed, new treatment
strategies that enhance efficacy are required. Here, we perform two independent genome-wide CRISPR screens to identify genetic determinants of CDK4/6 and endocrine therapy sensitivity. Genes
involved in oxidative stress and ferroptosis modulate sensitivity, with GPX4 as the top sensitiser in both screens. Depletion or inhibition of GPX4 increases sensitivity to palbociclib and
giredestrant, and their combination, in ER+ breast cancer models, with GPX4 null xenografts being highly sensitive to palbociclib. GPX4 perturbation additionally sensitises triple negative
breast cancer (TNBC) models to palbociclib. Palbociclib and giredestrant induced oxidative stress and disordered lipid metabolism, leading to a ferroptosis-sensitive state. Lipid
peroxidation is promoted by a peroxisome AGPAT3-dependent pathway in ER+ breast cancer models, rather than the classical ACSL4 pathway. Our data demonstrate that CDK4/6 and ER inhibition
creates vulnerability to ferroptosis induction, that could be exploited through combination with GPX4 inhibitors, to enhance sensitivity to the current therapies in breast cancer.
Cyclin D-dependent kinases 4 and 6 inhibitors (CDK4/6i) including palbociclib, ribociclib and abemaciclib in combination with endocrine therapy are the standard of care for patients with
estrogen receptor-positive (ER + ) and human epidermal growth factor receptor 2-negative breast cancer1,2,3,4,5. Inhibition of CDK4/6 and suppression of ER signalling each attenuate cell
proliferation by arresting cells in G1 of the cell cycle, and their combination enhances cell cycle arrest6. Despite the success of these treatments, the vast majority of patients progress
in the advanced setting, and many patients relapse despite adjuvant treatment. Therefore, novel approaches are required to overcome resistance and enhance sensitivity. Investigational oral
estrogen receptor antagonist and degraders, such as giredestrant, promote more effective suppression of ER and may be more effective than current endocrine therapies, especially in the
presence of ESR1 mutations7,8,9. Moreover, CDK4/6i have demonstrated limited activity in other types of breast cancer, such as triple negative breast cancer (TNBC), and therapies that
sensitise these cancers to CDK4/6i could have substantial utility.
Ferroptosis has been identified as an iron dependent non-apoptotic mechanism of cell death, mediated by accumulation of lipid peroxides that cause a rapid and unrepairable damage of the
plasma membrane10. Phospholipids (PL) that contain polyunsaturated fatty acids (PUFAs) are highly susceptible to peroxidation, and glutathione peroxidase 4 (GPX4) protects against
ferroptosis by catalysing the reduction of phospholipid and cholesterol hydroperoxides11,12,13. Various pathways regulate the membrane abundance of PUFA-PLs that are susceptible to
peroxidation, with ACSL4 and LPCAT3 generally being the key enzymes involved in this process14. Recently, peroxisomes have emerged as an additional regulator of ferroptosis, involved in the
synthesis PUFA ether-linked phospholipids (PUFA-ePLs) independently of ACSL4, with AGPS, FAR1 and AGPAT3 as key enzymes in this pathway15. Peroxidation of lipids can result from a
non-enzymatic reaction with alkoxyl radicals or hydroxyl radicals, or in an iron dependent Fenton-type reaction16.
To identify potential targets that enhance the efficacy of CDK4/6 inhibitors and endocrine therapy, we performed whole genome CRISPR/Cas9 suppressor screens, which unexpectedly identified
GPX4 depletion as the top hit sensitising palbociclib and giredestrant treated cells, with multiple other genes involved in ferroptosis modulating sensitivity. Here we show that GPX4
depletion sensitises multiple breast cancer models to CDK4/6 and ER inhibitors and investigate how these agents generate a ferroptosis vulnerable state.
To identify mechanisms of sensitivity and resistance to CDK4/6 inhibitors in ER+ breast cancer, we performed a negative selection genome-wide CRISPR/Cas9 mutagenesis screen using a human
sgRNA library containing 87,897 sgRNAs17 and doxycycline Cas9 inducible MCF-7 cell (MCF-7 iCas9; Fig. 1A). Following the sgRNA library transduction at a low MOI (MOI 0.3), cells were exposed
to doxycycline to induce Cas9 expression and gene mutagenesis, and then exposed to vehicle (DMSO) or palbociclib 500 nM (SF50) for two weeks. Deep sequencing was used to compare sgRNA
abundance in pre-treated (T0) and post-treated (T1) cell populations to identity genes that modified the response to palbociclib. In the vehicle controls we observed a significant depletion
of sgRNAs targeting commonly accepted “core essential genes”, suggesting the screen had sensitivity detecting genetic perturbations (Supplementary Fig. 1A). Moreover, the screen confirmed
previous findings. Hence, CRISPR-Cas9 targeting of the cell cycle inhibitors RB118,19,20, CDKN1A (p21), and FZR121, induced resistance to palbociclib (Fig. 1C, D). CRISPR-Cas9 targeting of
PI3K/AKT/mTOR growth pathway inhibitors PTEN19 and STK11 induced resistance, while loss of pathway activators GAB2 and LAMTOR5 increased sensitivity (Fig. 1C, D), supporting prior data
showing that activation of this pathway results in resistance to CDK4/6 inhibitors18,19,22,23,24. Our screen also confirmed that depleting cell cycle progression activators such as CDK2 and
CCNE1 increased sensitivity to palbociclib18,25,26. AMBRA1 that has been shown to lead to cyclin D1 stabilization and CDK4/6i resistance in a similar CRISPR screen27, was also a resistant
hit in our CRISPR screen (Fig. 1C), demonstrating the robustness of the screen.
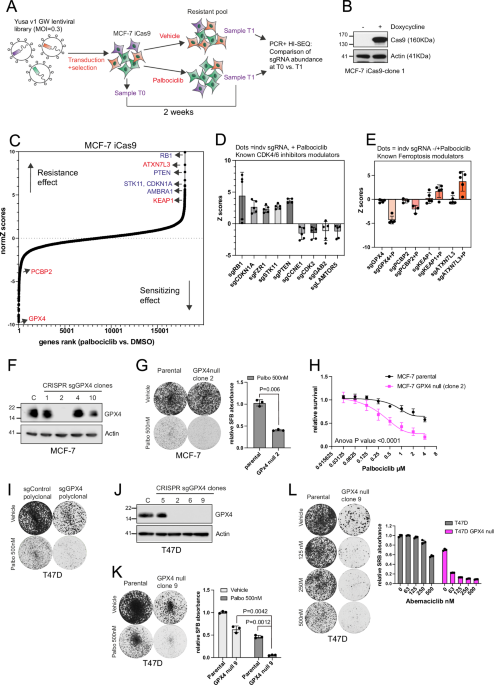
A Schematic illustrating a genome-wide palbociclib CRISPR/Cas9 screen in MCF-7 cell line. B Western blot analysis showing that MCF-7-iCas9 cells expresses Cas9 upon doxycycline. C Scatter
plot illustrating sgRNA pools normalised Z-score for palbociclib-sensitivity. Hits previously identified as modifiers of palbociclib sensitivity are highlighted in “blue”. Hits that are
ferroptosis modulators are highlighted in “red”. D Graph showing Z scores values for individual sgRNA (n = 4 or 5 different sgRNA per gene) of known hits involved in cell cycle or PI3K-mTOR
signalling, MCF-7 cells palbociclib-treated. E Graph showing Z scores values for individual sgRNA (n = 3 to 5 different sgRNA per gene) involved in ferroptosis that modulate palbociclib (P)
sensitivity, but do not influence untreated cells. F, J Western blot analysis showing GPX4 expression in MCF-7 and T47D parental (C) and GPX4 null clones developed by CRISPR Edit-R system. G
Clonogenics assay following 12 days treatment with 500 nM palbociclib or vehicle for the indicated cell lines. The graph represents relative SFB (sulforhodamine B) absorbance for the
palbociclib treated arm, mean with SD for three biological replicates (n = 3, paired t-test, P = 0.0066 for parental vs. GPX4null cells treated with palbociclib). H Dose–response survival
assays in MCF-7 parental and MCF-7-GPX4 null clone 2 cells exposed to palbociclib at the indicated concentration for 6 days. Graph shows relative survival, mean with SD for three biological
replicates (n = 3, Two-way Anova, P