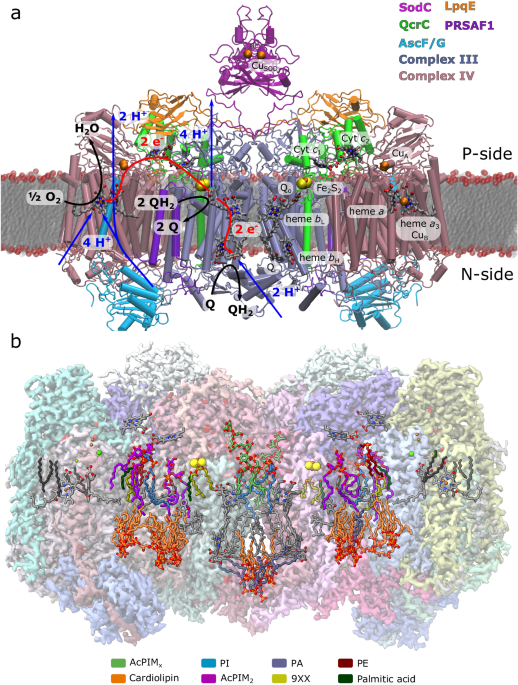
Long-range charge transfer mechanism of the iii2iv2 mycobacterial supercomplex
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Aerobic life is powered by membrane-bound redox enzymes that shuttle electrons to oxygen and transfer protons across a biological membrane. Structural studies suggest that these
energy-transducing enzymes operate as higher-order supercomplexes, but their functional role remains poorly understood and highly debated. Here we resolve the functional dynamics of the 0.7
MDa III2IV2 obligate supercomplex from _Mycobacterium smegmatis_, a close relative of _M. tuberculosis_, the causative agent of tuberculosis. By combining computational, biochemical, and
high-resolution (2.3 Å) cryo-electron microscopy experiments, we show how the mycobacterial supercomplex catalyses long-range charge transport from its menaquinol oxidation site to the
binuclear active site for oxygen reduction. Our data reveal proton and electron pathways responsible for the charge transfer reactions, mechanistic principles of the quinone catalysis, and
how unique molecular adaptations, water molecules, and lipid interactions enable the proton-coupled electron transfer (PCET) reactions. Our combined findings provide a mechanistic blueprint
of mycobacterial supercomplexes and a basis for developing drugs against pathogenic bacteria. SIMILAR CONTENT BEING VIEWED BY OTHERS STRUCTURAL BASIS FOR SAFE AND EFFICIENT ENERGY CONVERSION
IN A RESPIRATORY SUPERCOMPLEX Article Open access 27 January 2022 THE CRYO-EM STRUCTURE OF THE _BD_ OXIDASE FROM _M. TUBERCULOSIS_ REVEALS A UNIQUE STRUCTURAL FRAMEWORK AND ENABLES RATIONAL
DRUG DESIGN TO COMBAT TB Article Open access 02 September 2021 KEY ROLE OF QUINONE IN THE MECHANISM OF RESPIRATORY COMPLEX I Article Open access 18 August 2020 INTRODUCTION Aerobic
respiratory chains comprise membrane-bound redox enzymes that transfer electrons to oxygen and pump protons across a biological membrane1, powering the synthesis of ATP2. However, unlike the
mammalian electron transport chains (ETCs), which shuttle electrons from Complex I to Complex IV, bacteria employ highly branched ETCs that vary in their composition and operational mode
depending on the external conditions1. The enzymes responsible for the ETC can operate as higher-order supercomplexes (SCs)3,4,5, although their functional role remains elusive4,6,7,8,9. The
architecture of several SCs with different stoichiometries of Complexes I, II, III, and IV, were recently resolved10,11,12,13,14,15,16,17,18,19,20,21, including the mycobacterial III2IV2 SC
from _M. smegmatis_10,11,12,13,14 and related species15,16,22 that forms an obligate assembly operating only as a single functional unit. Mycobacteria comprise several pathogens that cause
serious diseases such as tuberculosis, with major impact on global health23. Understanding the bioenergetic principles of this mycobacterial SC could thus provide new avenues for developing
drugs against emerging multi-resistant bacteria. Recent cryo-electron microscopy (cryo-EM) studies of the III2IV2 SC from _M. smegmatis_ revealed a 0.7 MDa dimeric protein complex,
comprising up to 26 subunits and stabilised by cardiolipin at the dimer interface10,12,13 (Supplementary Fig. 1a). The structures also resolved partial density for menaquinone molecules in
non-canonical binding sites (here Qo2 and Qo1b), and inhibitors in the Qo1a site, including Q203 and TB4712,13, currently used as drugs against tuberculosis. The mycobacterial SCs harbour
several adaptations that could be of functional relevance. _M. smegmatis_ lacks genes for a soluble cytochrome _c_ and employs instead the QcrC subunit to shuttle electrons between Complexes
III and IV (Fig. 1), thus making the SC assembly necessary for its proper function (Supplementary Fig. 1h). The SC also features a C-type superoxide dismutase (SodC) and subunit LpqE, both
of which are anchored to the membrane by lipid modifications (Supplementary Fig. 1c, d). The SC was observed in both symmetric11,13 and asymmetric assemblies10,12 with respect to the QcrC
module, establishing contacts with either both or one of the Complex III and IV domains, respectively. The symmetric model features LpqE, as well as two closed QcrCs that bridge between the
complexes, while the asymmetric model lacks LpqE and results in an open QcrC with a disconnected electron transfer pathway (Supplementary Fig. 1a, b)10,11. While dissociation of LpqE is
likely to arise from purification artefacts, it cannot be fully excluded that switching into an asymmetric state could also provide a possible regulatory function. Despite the structural
differences that are likely to tune the energetics of the charge transfer process (see below), the conserved structural elements5 of the mycobacterial SC, suggest that it could utilise
overall similar charge transfer pathways as Complex III and IV1,24,25,26 variants, not found in obligate bacterial SCs (from here on referred to as canonical Complexes III and IV, Fig. 1).
In this regard, the Complex III module of the SC could employ the well-established Q-cycle mechanism27,28, in which the Qo site bifurcates the electrons from menaquinol (instead of ubiquinol
used in the canonical Complex III) to the Rieske FeS centre and further to the QcrC domain, whereas the other electron transfer branch shuttles the electrons via the low and high potential
hemes _b_L and _b_H to the Qi site, where another quinone is stepwise reduced to quinol (Fig. 1). The quinol oxidation at Qo releases the protons to the positively-charged (P-side) of the
membrane, whilst quinone reduction at the Qi site results in proton uptake from the N-side. However, due to the unique mycobacterial subunits, the proton pathways likely follow different
routes as those revealed in prior structures of the canonical isoforms29,30. The complete turnover of the Complex III module thus involves two quinol oxidation steps at the Qo site and one
quinone reduction at the Qi site that establishes a proton gradient across the membrane. The electrons released at the Qo site are transferred via the QcrC domain (cyt _c_1_c_2) to CuA of
Complex IV, without a soluble cyt _c_10,11,12,13,14 (Supplementary Fig. 1h), which also implies that the mycobacterial SC catalyses electron bifurcation without the characteristic motion of
the Rieske domain31,32. In this regard, experiments33,34,35 and recent DFT studies36 revealed insight in the proton-coupled electron transfer (PCET) steps between the quinol and the FeS
Rieske centre for the canonical Complex III, while the PCET mechanism in the mycobacterial SC remains poorly understood. The electrons then continue from the QcrC domain and CuA, further to
heme _a_ and the binuclear heme _a_3/CuB centre (BNC), responsible for oxygen reduction to water24. Prior data on Complex IV24,37 show that the inter-heme electron transfer couples to proton
uptake via the D- and K-channels (named after the conserved Asp115 and Lys340 in _M. smegmatis_ numbering) that also have modular adaptations as compared to the canonical enzyme. These
channels shuttle the protons both to the active site and across the membrane24. The Complex IV domain of the SC is expected to operate as a redox-driven proton pump, in contrast to the
Complex III domain that generates _pmf_ via the redox-loop mechanism (cf. refs. 1,26 for discussion on different proton translocation mechanisms). To address the functional dynamics
responsible for this fascinating long-range charge transfer process in the mycobacterial III2IV2 SC, we integrate here structural, functional, and computational methods. More specifically,
we aim to address principles of quinone binding in the SC, the mechanism and energetics of the electron bifurcation process, how the formation of functional water wires enables the catalysis
and proton release, and how modular adaptions in the catalytic site could tune the overall energetics. To this end, we combined large-scale atomistic molecular dynamics (MD) simulations and
hybrid quantum/classical (QM/MM) free energy simulations with high-resolution cryo-EM analysis and biochemical activity assays, allowing us to study the link between catalysis and
conformational dynamics. Our high-resolution structures (2.3 Å, 2.8 Å) together with multi-scale simulations and functional assays resolve unique proton pathways, quinone binding sites, and
position of lipid molecules of the mycobacterial SC. These findings establish structural and mechanistic understanding of the PCET energetics and redox tuning effects, and they explain how
the mycobacterial Complex III catalyses electron bifurcation. Importantly, the differences to the well-studied canonical complexes (cf. refs. 1,24,25,26,32) provide an important basis for
drug design against pathogenic bacteria. RESULTS GLOBAL DYNAMICS AND PROTON PATHWAYS OF THE SC To probe the functional dynamics of the _M. smegmatis_ III2IV2 SC, we constructed a symmetric
model with respect to the QcrC subunit based on cryo-EM data (see Methods). The SC model was embedded in a lipid membrane, and modelled in different redox and protonation states, with the Q
species modelled in the canonical (Qo1a) and non-canonical sites around Qo (Qo1b, Qo2) (Fig. 2). Moreover we modelled a menaquinone in the Qi site, and the cofactors of Complex IV in their
oxidised states24 (see Methods, Supplementary Figs. 1 and 2, Tables 1–3, and Methods for detailed simulation models, redox and protonation states). The total system comprised around 1
million atoms, which we explored by ca. 10 μs atomistic MD simulations in different states along the reaction cycle (Supplementary Table 1, Movie 1). While the focus here is on the symmetric
SC, we also conducted MD simulations on an asymmetric SC model (see above, Methods) to probe how the possible open QrcC conformation could affect interactions with SodC and the electron
transfer dynamics. To experimentally validate our findings, we performed cryo-EM experiments, where we refined a glyco-diosgenin (GDN) detergent-solubilised, symmetric SC to an overall
resolution of 2.3 Å, and a local resolution of 2.0 Å for the core parts, and another dataset of the same state, to 2.8 Å global resolution (to 2.5 Å at the core), enabling us to
independently determine the position of functionally central and previously unresolved water molecules that are key for the proton transfer reactions. We focus here on the higher resolution
dataset, which contains more details, while the other dataset shows overall similar results and serves as an independent validation of our model. We also characterised the quinol
oxidation-O2 reduction activity by biochemical assays that we compared with kinetic network models of the long-range charge transfer process. The global dynamics extracted from the MD
simulations of the SC closely resemble the variation in the local resolution in both our current and prior cryo-EM maps (Supplementary Fig. 3). The lipid-anchored SodC module forms the most
dynamic part of the SC in the MD simulations, consistent with the blurred density observed in our cryo-EM data (Supplementary Fig. 4). The SodC samples distances in the range of ca. 40–80 Å
from the QcrC domain in the symmetric model, and ca. 25–50 Å for the asymmetric model (Supplementary Figs. 1, 4b). Other highly dynamic regions include QcrC (open conformation), AscF/G
(previously MSMEG 4692/469322), and LpqE, whereas the dimeric SC interface, formed by cardiolipin molecules (see below), remains highly stable during the simulations (Supplementary Figs. 1
and 3). The SC becomes highly hydrated in functionally important regions during the MD simulations, leading to a total influx of around 900 water molecules in the buried parts of the protein
(Supplementary Fig. 5) that could not be resolved in prior cryo-EM studies of the _M. smegmatis_ SC (but cf. ref. 15). However, our current high-resolution cryo-EM analysis revealed around
600 functionally relevant water molecules (at 2.3 Å, ca. 200 for the 2.8 Å map) that compare well with the data from our MD simulations (see below). More specifically, our cryo-EM data show
20–30 water molecules between the Qo1a site and QcrC that are in excellent agreement with our MD simulations (Fig. 3c, Supplementary Figs. 5 and 6). These water molecules establish a
hydrogen-bonded pathway from the Qo1a site via Tyr159, Asp302, and His110 to the P-side, whereas the other branch connects to the P-side via His355, Asp309, and Arg313 (Fig. 3a, b,
Supplementary Fig. 6a). From the N-side of the membrane, we also observe a putative proton pathway from Lys260 and Glu44 to Qi in both the MD simulations and the cryo-EM data (Fig. 3d, i).
On the Complex IV side of the SC, our combined MD and cryo-EM data show hydrogen-bonded water arrays along the D- and K-channels that support the proton uptake from the N-side of the
membrane to Glu266 (via the D-channel) or via Lys340 to Tyr268 of the BNC (via the K-channel) (Fig. 4d–f)38,39 (cf. also refs. 15,40,41). Glu266, the terminal residue of the D-channel
samples both downward and upward conformational states (Fig. 4b) in the MD simulations that favour contacts with the respective D-channel and a putative proton-loading site (PLS) near the
propionates of heme _a_3, that transiently stores protons during proton pumping across the membrane in canonical Complex IV (cf. ref. 42). When Glu266 is in the upward conformation, the
non-polar cavity next to the BNC occupies around 6 water molecules during the MD simulations, with Glu266 forming proton wires both to the propionate groups of heme _a_3 or to the BNC (Fig.
4a)24,40,41,42,43 that could direct the protons for pumping or to be consumed in the reduction of O2 to H2O, respectively40,41. In contrast, the downward conformation is less hydrated in the
MD simulations and lacks clear proton pathways between Glu266 and the BNC. Our cryo-EM data show a well-resolved density of the entire sidechain of Glu266 in the downward conformation
(Supplementary Fig. 7c), possibly due to the protonated form of the residue, and further supporting that the residue has a high p_K_a value, consistent with its function as a proton shuttle
(cf. refs. 42,44). We further note that while Glu266 samples both conformations in the MD simulations, the downward conformation is energetically preferred, consistent with the lack of
cryo-EM density for the upward conformation. From the propionate region of heme _a_3, we observe pathways that support proton release from the Mg2+ site to the bulk P-side of the membrane,
also in line with prior data on the proton release pathways in canonical Complex IVs43,45 (Fig. 4c, i, see section: Analysis of the high-resolution cryo-EM structure). QUINONE BINDING MODES
ENABLING CATALYSIS To obtain insight into the long-range electron transfer process, we next studied binding of menaquinol around the Qo sites (Fig. 2a–d). The Qo1a site corresponds to the
canonical Qo site in cytochrome _bc_1 with previously resolved inhibitors at this location13,15,46 (cf. also ref. 32, see Methods for details of modelling menaquinol binding to this site).
In our cryo-EM data, we observe densities for menaquinone only in the Qo1b and Qo2 sites, consistent with previous studies10,11,13,15,22, although their functional roles for catalysis have
remained unclear. Our MD simulations suggest that menaquinol is stabilised in the Qo1a site by His355 and Asp309, and by water-mediated contacts with Tyr159, which in turn interacts with
Asp302 of the characteristic PDFY motif47,48 (Figs. 2b, 3a–c, Supplementary Fig. 9). Asp302 establishes an ion-pair with His110, the conformation of which modulates the binding affinity of
the substrate (Supplementary Fig. 9e). The water wires leading from the menaquinol to His110/Asp302 and Asp309/Arg313 are connected to the P-side bulk, forming possible proton exit pathways
that are also well-resolved in our cryo-EM data, with water positions closely matching those observed in the MD simulations (Fig. 3a–c, Supplementary Figs. 6, 10). In the Qo1b site, the
menaquinol is at ca. 10 Å edge-to-edge distance from the FeS Rieske centre and somewhat further (12–17 Å) from heme _b_L (Fig. 2e). The menaquinol headgroup is stabilised by π-stacking
interactions with Phe153, and non-polar contacts with, e.g. Met305 and Met337 (Fig. 2c, Supplementary Fig. 8a, b). In the Qo1a site the menaquinol is closer to the redox centres, ca. 5 Å
edge-to-edge distance from the FeS Rieske centre, and around 12 Å from heme _b_L (Fig. 2b, e). Interestingly, in some simulations initiated from the Qo1a site, we observe a transient motion
of the quinol towards the Qo1b site, suggesting that these regions are dynamically exchangeable, with comparable binding energies (Supplementary Fig. 8h–j). Our MD simulations suggest that
the menaquinol is dynamically flexible at the Qo2 site, sampling several binding poses with a broad edge-to-edge distance distribution at ca. 20 Å between the quinone and heme _b_L/FeS
Rieske centre (Fig. 2e). The menaquinol forms π-stacking / hydrogen-bonding interactions with Trp276, Thr388, Phe303, and Tyr384 (Fig. 2d), consistent with the well-resolved density in the
cryo-EM maps (Fig. 3g). Despite the stable binding mode (Supplementary Fig. 8h–j), we do not observe residues that could support the proton-coupled oxidation of the menaquinol, suggesting
that Qo2 is not directly employed for catalysis. Moreover, in contrast to the exchangeable Qo1a/b sites, we could not observe substrate tunnels connecting Qo2 with the Qo1a/b sites, despite
similar binding energies amongst the sites in the MD simulations (Supplementary Fig. 8h–j). PROTON-COUPLED ELECTRON TRANSFER DYNAMICS IN THE QO SITE We next addressed the mechanism of the
PCET reactions during the menaquinol oxidation by QM/MM free energy simulations, which allowed us to study the energetics of bond-breaking and formation at a quantum mechanical level
(Methods, Supplementary Fig. 11). These simulations show that the proton transfer from the menaquinol to His355 in the Qo1a site takes place by a concerted PCET process, where the proton
moves along the menaquinol-His355 hydrogen-bond and the electron is transferred from the aromatic ring of the quinol directly to the (His355-ligated) ferric iron of the FeS Rieske centre
(Fig. 5a, e, Supplementary Fig. 11, Movie 2). The PCET reaction has a free energy barrier of around 5 kcal mol−1 (Fig. 5c) and the process is weakly exergonic, consistent with redox
mid-point potentials of the menaquinol49 (_E_m,7exp = −80 mV) and the FeS centre (_E_m,7exp = +160 mV, Δ_G_exp = −5.5 kcal mol−1; Δ_G_QM/MM = −4.6 kcal mol−1), (cf. also refs. 36,50,
Supplementary Table 4). After the initial H+/e− transfer step, the resulting semiquinone (QH•) deprotonates via a water-bridge contact via Tyr159 to Asp302 (Fig. 5b, d, Supplementary Fig.
11, Movie 3). The second PCET process leads to a Grotthuss-type proton transfer reaction, with a barrier of +11 kcal mol-1 (rate of 14 μs−1) that triggers an electron transfer to heme _b_L
(Fig. 5d, Supplementary Fig. 11). We note that the Qo1b site is unlikely to support a similar reaction due to the long distance between the Q and His355/Tyr159/Asp30 (see also Supplementary
Table 4, Fig. 12). As usual in biological electron bifurcations51,52,53,54, this endergonic charge transfer step could be driven by the initial exergonic PCET step (Fig. 5d). The energetics
suggests that the second PCET step is rate-limited by the proton transfer followed by a rapid non-adiabatic electron transfer reaction. The proton is directed from this PCET process to the
His110/Asp302 ion-pair, which could hold the quinol proton until the electron has been transferred to the heme _b_H / Qi site. In this regard, electrostatics calculations (Fig. 6f) suggest
that the oxidation of heme _b_L lowers the proton affinity of the His110/Asp302 cluster, and favours the proton release to the P-side bulk via the water arrays described above (Fig. 3a, c,
Supplementary Fig. 6a). These local PCET reactions further couple to the energetics of quinol binding and quinone release, whilst the overall process is driven by the exergonic oxygen
reduction reaction at the BNC (see Supplementary Fig. 24 for schematic free energy profile). Taken together, these findings suggest that the PCET-driven quinol oxidation is catalysed by the
Qo1a site. LONG-RANGE ELECTRON TRANSFER KINETICS To probe the long-range charge transfer pathways within the SC, we next developed a kinetic network model based on our multi-scale
simulations, experimental redox potentials, reorganisation energies26,49, as well as kinetic measurements55,56 (see Methods, Supplementary Fig. 12). While the models explicitly account for
electron transfer reactions, the quinol/quinone binding/unbinding (in Complex III) and water formation (in Complex IV) are implicitly accounted for by modelling the associated steps as
irreversible. Our model suggests that the symmetric SC is in complete charge transfer contact from the Qo1a site of Complex III to the BNC of Complex IV, with an overall PCET rate within the
SC of ca. 88 s−1 (Fig. 6d), which is roughly 40 times slower relative to the canonical _aa_3-type Complex IV57,58, but compares very well with our experimental SC activity of around 90 s−1
of GDN-solubilized III2IV2 SC (Fig. 6c, e) (cf. also ref. 13). In this regard, our model accounts for the proton-coupled reduction of the BNC41 and the experimentally measured proton uptake
rate in the SC Complex IV of ~4 ms55,56 that tune the apparent redox potential of the BNC (see Supplementary Information). Without such redox gates, the model predicts a pure electron
transfer flux in the SC of 1000–2000 s−1, i.e. an order of magnitude faster rate as compared to our experimental observations (Fig. 6a, Supplementary Fig. 13, Table 4). Interestingly,
however, our kinetic model suggests that the electron initially reaches a pre-steady state, with the electron distributing on heme _a_, CuA, and heme _c_2 centres until the proton uptake
from the D-channel tunes the redox midpoint potential of heme _a_3/CuB that completes the reaction (Fig. 6b). The complete turnover of the canonical Complex III takes place with a rate of
ca. 120–300 s−159,60,61, whilst our kinetic models and activity data suggests that mycobacterial Complex III is not rate-limiting for the long-range charge transfer process, although it has
not been possible to experimentally determine uncoupled activities and potential rate-limiting steps in the mycobacterial Complex III. Overall, these findings suggest that the proton uptake
is rate-limiting for the overall long-range PCET network within the SC, with the observed turnover determined by the proton uptake to Complex IV. To further address the electron transfer
within the SC dimer, we analysed our MD simulations of the asymmetric SC, where the QcrC interface remains in an open conformation (Fig. 1a, Supplementary Fig. 1), consistent with previous
structural data10,12. This, in turn, prevents electron transfer on physiologically relevant timescales on one side of the dimer, whilst we observe a similar electron transfer flux as in the
symmetric model in the closed protomer (Supplementary Fig. 13d). Our models also predict a slow electron transfer rate relative to the overall turnover62 (but cf. also ref. 34) between the
Complex III protomers via the heme _b_L—heme _b_L bridge (ca. 10 s−1, Supplementary Fig. 13e, f). Purification of the SC with _n_-dodecyl-β-D-maltoside (DDM) leads to dissociation of LpqE
from the SC, which is supported by structural data showing only a weak density for the subunit. Small amounts of the LpqE subunit remain in the sample or associated to the SC (Fig. 6e, see
also ref. 12), and could explain why the DDM preparation shows only around 30% reduction of the SC activity (66 s−1) relative to the GDN-solubilized SC, where all subunits are present (Fig.
6c), instead of a 50% reduction as may be expected, if one electron transfer branch would be completely blocked. The reduced activity is likely to arise from a significant amount of open
QcrC conformations, consistent with previous structural data10 and our kinetic models (Fig. 6a, b). On the acceptor side of Complex III, we observe a subtle motion of the Qi quinone from the
resolved binding position to a site, ca. 4 Å further from heme _b_L, where the quinone forms a π-stacking interaction with Trp231 in the MD simulations (Fig. 3d, e). The heme _b_L—heme
_b_H—Qi network favours rapid electron transfer (103–104 s−1, Fig. 6a), with our QM/MM simulations (Supplementary Fig. 14) further suggesting that Lys260/Glu44 or Lys32 could function as
local proton donors upon the quinone reduction (Supplementary Fig. 14, Movie 4), following uptake of the protons from the N-side via the nearby cardiolipin gate (see below, and cf. also
refs. 30,63). In this regard, our simulations suggest that proton transfer may only occur after the Qi species has been fully reduced (Supplementary Fig. 14, Movies 4, 5). Our cryo-EM data
revealed a well-defined density of a cardiolipin molecule and two water molecules near the Qi site that support the positioning of the Qi headgroup and the putative cardiolipin
lipid-mediated proton uptake route (Fig. 3i). These pathways are readily accessible from the N-side bulk, and therefore not expected to be rate-limiting for the overall long-range charge
transfer process. ANALYSIS OF THE HIGH-RESOLUTION CRYO-EM STRUCTURES The two cryo-EM datasets of the _M. smegmatis_ III2IV2 SC allowed us to gain experimental insight into the SC function
and validate our computational findings (Fig. 1b, Supplementary Figs. 15, 16). The refined structures revealed a symmetric SC, comprising 22 main subunits, including LpqE. The data allowed
us to assign densities of 614 water molecules (in the 2.3 Å map, and 249 in the 2.8 Å map, Supplementary Fig. 10), many of which are located around key functional regions in Complexes III
and IV, as discussed above. The two datasets are consistent with each other, resolving water molecules at the same positions in several sites, but with significantly higher statistical
certainty in the 2.3 Å resolution map. Importantly, the higher (2.3 Å) resolution map allowed us to further assign functionally central water molecules near CuB, as well as in the D- and
K-channels. These experimentally determined water positions are in excellent overall agreement with the hydration dynamics independently predicted by our MD simulations, supporting the
complementarity between the approaches, but not observed in the 2.8 Å map due to possible averaging effects. The D-channel shows 10 water molecules connecting Asp115 with Glu266 (Fig. 4e,
j), supporting the proton pathway in the D-channel. The non-polar cavity next to the BNC that establishes a proton wire between Glu266 and CuB in the MD simulations (Fig. 4a), shows a weak
cryo-EM density for a water molecule near CuB (Fig. 4h, inset). The K-channel harbours five water molecules at the channel entrance close to Glu539, as well as water molecules next to Lys340
(Fig. 4k), between Thr337, Tyr268, and the BNC that could support the proton uptake. Overall, more water is present in the D-channel as compared to the K-channel. This could, at least in
part, arise from the larger volume of the D-channel (Supplementary Fig. 18d), but also from the higher water mobility in the K-channel, as suggested by the MD simulations (Supplementary Fig.
18e). We note that prior high-resolution structures (PDB ID: 7COH64, 7ATE65, 7AU665) of the canonical Complex IV support similar hydration levels. Interestingly, key residues along a
previously proposed H-channel66 are also present in the _M. smegmatis_ SC, but both our cryo-EM data and MD simulations show low hydration of the region, suggesting that it is unlikely to
conduct protons (cf. also ref. 67). The heme _a_3 iron of the BNC has a spherical density, and the shape of the porphyrin ring is well-resolved in the 2.3 Å map (Fig. 4h, Supplementary Fig.
7e–j). We could assign a Fe-Cu distance of 4.4 Å, and the continuous density connecting the metals could fit, e.g. a hydroxyl and a weak water ligand, based on QM/MM calculations, although
assignment of other ligands is also possible (Fig. 4h, Supplementary Fig. 7e–j) (cf. also65,68,69). The characteristic His264-Tyr268 crosslink is well-resolved (Fig. 4h), with a refined C–N
bond distance of 1.43 Å (based on QM/MM), and the heme _a_3 farnesyl further stabilising the phenol group of Tyr268. The non-polar cavity is occupied by one water molecule with a rather low
cryo-EM density (Fig. 4h), possibly due to the downward conformation of Glu266, which also leads to a low hydration level in the MD simulations. We further note that high-resolution x-ray
and cryo-EM structures of the canonical Complex IV (PDB IDs: 7COH64, 7ATE65, 7AU665) show similarly a dry cavity, together with Glu266 oriented towards the D-channel. The propionates of heme
_a_3 comprise several water molecules that connect to the P-side bulk (Fig. 4i, c, Supplementary Fig. 6b, 10). We further resolved a Ca2+ ion in pentagonal bipyramidal coordination around
12 Å (edge-to-_edge_) from heme _a_ (Supplementary Fig. 7b, d), in a position that could affect the redox properties of heme _a_, and/or the proton release along the exit pathway (cf. refs.
70,71,72). On the Complex III side of the SC, we observe ten menaquinone molecules (five per monomer) at five different positions. The canonical Qo1a site is empty (cf. also refs. 10,12,13),
but a menaquinone occupies the Qo1b site (Fig. 3f). Another menaquinone binds next to Trp276 in the Qo2 site at the interface of Complexes III and IV (Fig. 3g), with an additional site
close to the P-side (Fig. 1b), as also suggested in previous studies10,12,13. We also observe density corresponding to two menaquinones at the N-side of the membrane, one at the canonical Qi
site, and one in another position, Qi,2, ca. 10 Å away from the former (see Fig. 3h), with possible substrate queuing or regulatory functions. The former site allows modelling of the
quinone headgroup in alternate positions, with the weaker density suggesting that the Qi,2 site has a lower affinity (cf. also PDB ID: 7E1V13). Due to the large distance from the residues
supporting PCET during the Qi reduction, this accessory site is, however, unlikely to be involved in catalysis. We note that the quinones at the Qi site show partially overlapping densities,
which could arise from an ensemble average of different binding modes. Our resolved SC structures show multiple lipid-binding sites (Fig. 1b, Supplementary Fig. 17), including cardiolipin
molecules on the N-side at the interface between the Complexes III and IV that could have a central structural role, as also suggested for other SCs73. We find further cardiolipins near the
Qi site that are in different positions from those previously described for the mammalian Complex III63, in place of which the _M. smegmatis_ SC harbours the QcrC helix. One of these
cardiolipins establishes a contact via water molecules and Lys32 to Qi and could catalyse the proton uptake from the N-side (Fig. 3i). The interface between Complexes III and IV contains
four cardiolipin molecules on each side, three cardiolipins between PRSAF1 and Complex IV, and one cardiolipin next to subunit III of Complex IV (Fig. 1b). Two acyl
phosphatidylinositol-hexamannoside (AcPIM6) molecules, and four phosphatidylinositol (PI) lipid molecules are located near Qo1a/b, the QcrB/PRSAF1 and the QcrA/QcrB interfaces (Fig. 1b,
Supplementary Fig. 17), with the AcPIM2 molecules next to the QcrC helix (Supplementary Fig. 7a), serving a possible structural role. The PIM lipids are characteristic for mycobacterial
membranes74 and likely to affect the biophysical properties of the bacterial membrane and/or the function of the SC. The Ac-PIMs are resolved on the P-side of the SC, consistent with
previous assignments suggesting that mycobacterial membranes have an asymmetric lipid distribution with the P-side leaflet enriched by PIM6 lipids74. DISCUSSION Our integrative
structure-function approach revealed key water structure, lipid interactions, and modular adaptations that govern the long-range PCET dynamics in the III2IV2 SC from _M. smegmatis_. Our
combined findings show that the SC establishes an electronic wiring between the Qo1a site of Complex III and the active site of Complex IV, leading to a turnover rate of ~90 s−1. Our
large-scale MD simulations and high-resolution cryo-EM structures, resolved over 600 water molecules, which enable the proton release from the Qo1a site, proton uptake to the Qi site, as
well as proton transfer across both the D- and K-channels to the oxygen reduction site and across the membrane. These proton transfer reactions support the redox-loop and redox-driven proton
pumping mechanisms of the Complex III and IV modules, respectively. On the Complex III side of the SC, the modular adaptations at the Qo1a site have important redox tuning effects that
modulate the PCET reactions both to the FeS Rieske centre and to heme _b_L (Fig. 7). The Qo1a site catalytically supports these reactions, whereas that unique Qo1b site (Fig. 2e) may
represent a transient substrate docking position, which can exchange with the Qo1a site. In contrast, the Qo2 site is not structurally connected to Qo1a, and the site lacks residues that
could support the menaquinol oxidation. The Qo1b and Qo2 sites are resolved in our high-resolution cryo-EM structures, and are consistent with previous data10,12,13, whilst the properties of
the Qo1a site were explored by multi-scale simulations. The water wires observed in our cryo-EM structures and MD simulations support the proton release upon the menaquinol oxidation via
the QcrB/QcrC interface to the P-side of the membrane (Figs. 3a, c and 7) (cf. also ref. 15). The mycobacterial Qo site could have evolved to account for the shifted redox properties of
menaquinol relative to ubiquinone (Fig. 3f), as suggested by in silico mutagenesis and electrostatic models (see Supplementary Information). These calculations suggest that Asp309 has an
important redox tuning effect that favours the PCET reaction: in the D309N variant, the redox potential of the FeS Rieske centre is downshifted by ca. 100 mV and heme _b_L is upshifted by 50
mV relative to the WT model (Fig. 6f, Supplementary Table 5). Protonation of His355 increases the redox potential of the FeS Rieske centre by ca. 190 mV, leading to an effective
proton-coupled redox potential of +120 mV (Fig. 6f). In this regard, Asp309 could help in establishing a downhill electron transfer between semiquinone and heme _b_L, a process that is more
favoured with the ubiquinol (+90 mV). We suggest that these adaptations could be important for the mycobacterial Complex III, where the Rieske domain does not undergo a conformational change
that kinetically gates the electron transfer reaction75. The redox tuning effects around Qo1a could thus be critical for ensuring a high forward electron flux. Moreover, the quinone motion
from Qo1a to Qo1b could provide additional entropic effects to compensate in part for the non-mobile Rieske protein. The electron transfer between the quinone and heme _b_L is linked to a
proton transfer from the semiquinone (QH•) to Asp302 (Fig. 5b, d), which is adjacent to His110. The His110/Asp302 ion-pair undergoes conformational changes in the MD simulations and could
modulate the energetics of the proton release to the P-side bulk. Protonation of the His110/Asp302 also has a redox tuning effect on heme _b_L, with the proton release to the P-side expected
to downshift the redox potential of heme _b_L that could favour the forward electron transfer (Fig. 7). The tuberculosis drug molecules, Q203 and TB47 interact with Asp309 (Glu314 in _M.
tuberculosis_) and His355 (His375 in _M. tuberculosis_)12,13 that could be further targeted in rational drug design, e.g. by introducing functional groups on the inhibitor that form specific
interactions with these residues. In the Qi site, we also observed changes relative to the canonical Complex III, specifically a π-stacking interaction between the menaquinone and Trp231,
whereas Lys32 and/or Lys260 could serve as proton donors in the Qi reduction, catalysing proton uptake via the bound cardiolipin molecule (Fig. 3d, i, Supplementary Fig. 14) (cf. also refs.
30,63). On the other side of the electron bifurcation pathway, our data suggested that the electrons are transferred from the Rieske FeS centre via the two heme _c_ cofactors of QcrC to
Complex IV that form the rate-limiting step for the charge transfer due the large distance between the donor-acceptor sites, consistent with previous experiments55,56. By accounting for the
experimental proton uptake rate55, we could predict an overall turnover rate that is in excellent agreement with our experimental activity data. These findings support that the slow proton
uptake in Complex IV module of the SC is rate-limiting for the overall long-range PCET process. Our data also showed that the mycobacterial-specific SodC subunit is dynamically highly
flexible, forming contacts with the QcrC domain, albeit the electron transfer from the Cu of SodC to the QcrC is expected to be rather slow based on the MD-sampled conformations. Although
not supported by our current experimental data, it is interesting to speculate whether such putative electron transfer routes could provide an alternative electron input channel for the SC
under specific conditions, and a detoxification mechanism for the mycobacterial SC to scavenge reactive oxygen species created, e.g. by human immune cells (cf. also refs. 10,76,77). On the
Complex IV side of the SC, the D-channel establishes a proton wire from Asp115 to Glu266, and further via a putative PLS near heme _a_3 and to the P-side bulk (Fig. 4c). Glu266 sampled both
upward and downward conformations in our MD simulations, enabling contacts with the active site/the PLS and the D-channel, whilst our high-resolution cryo-EM structure support only the
downward conformations of the residue (but cf. refs. 15,41,42). Our MD simulations together with structural analysis55 suggested that the D-channel is partially blocked by the QcrB loop
(Fig. 4g), with several protonatable residues that could control the proton uptake into the channel (Supplementary Fig. 18a). The slow water exchange at this site (Supplementary Fig. 18b)
could affect the proton uptake from the N-side and rationalise the pH-independent P → O transition in the _M. smegmatis_ SC55. We suggest that the protonatable groups of the QcrB loop
function as a proton-collecting antenna that renders the SC less sensitive to environmental changes. Our data also supported several water molecules in the K-channel that are involved in
proton uptake to the active site. Despite key modular adaptations, the Complex IV domain of the SC is likely to be governed by overall similar functional principles as the canonical
heme-copper oxidases, but establishing an overall rate-limiting step for the long-range charge transfer process. Moreover, the mycobacterial-specific residues and proton wires that control
the primary PCET reactions in the Qo site of Complex III, provide a basis for site-directed mutagenesis experiments and establish principles for the rational drug design of inhibitors of
mycobacterial SCs. In conclusion, we have described here mechanistic principles of the obligate III2IV2 SC using large-scale molecular simulations in combination with structural experiments
and activity measurements. Our simulations show that quinol can bind in the canonical Qo1a site, where it interacts with the nearby residues His355 and Tyr159. The binding mode is consistent
with previous inhibitor-bound forms of the canonical Complex IIIs46, whereas the unique Qo1b site is exchangeable with the former, and could provide a transient substrate docking site. Our
QM/MM free energy calculations predict that the Qo1a site enables an exergonic PCET reaction from quinol to the FeS centre followed by a second, endergonic PCET towards the heme _b_L. In
this regard, modular adaptations of specific residues in the Qo1a site tune the energetics of these redox reactions, which could enable mycobacteria to use menaquinol as a substrate instead
of ubiquinol and support electron transfer without the characteristic Rieske motion as in canonical Complex IIIs. Our MD simulations together with our structural data show functional proton
wires in central sites of the SC, including the Qo1a site and Qi sites in Complex III, as well as around the active binuclear site and the D- and K-channels in Complex IV. Our predicted
electron transfer rates in the SC are in good agreement with measured activities and further support that proton uptake via the D-channel is the rate-limiting step for the long-range charge
transfer process. Taken together, our combined findings support previous data on the well-studied Complexes III and IV24,26,32 but also highlight key differences in the quinone binding,
proton pathways, and redox tuning principles that provide a basis for further mechanistic studies and for developing drug molecules against mycobacteria. METHODS MOLECULAR MODELS The III2IV2
supercomplex of _M. smegmatis_ was modelled based on the cryo-EM models (PDB ID:6ADQ11 and PDB ID: 6HWH10) (see Supplementary Fig. 1, Table 2). A SodC dimer was homology modelled based on a
high-resolution x-ray structure of the protein from _Mycobacterium tuberculosis_ (PDB ID; 1PZS78) using SWISS-model79 and docked into the blurred cryo-EM density via the lipid membrane
anchor, which we covalently linked to Cys21SodC, as also observed in previous structures10,11,80,81. Similarly, Cys24 of subunit LpqE was also linked to a lipid membrane anchor. A menaquinol
was modelled in three putative positions (Qo1a, Qo1b and Qo2). The Qo1b and Qo2 sites were modelled based on the partial density observed in refs. 10,11,82, whilst the Qo1a position was
modelled based on the resolved positions for quinone (PDB ID: 6Q9E) from ref. 16 and stigmatelin (PDB ID: 1PP9) from ref. 83 To this end, we applied a harmonic force (_k_ = 20 kcal mol−1
Å−2) between the quinone in the Qo1b site and H355A and pulled the quinone gradually until it reached a hydrogen-bonding distance with H355A (3.5 Å between the nearest quinone oxygen and the
NE2 of H355A). During the minimisation, the protein backbone was fixed with residues only in QcrB allowed to relax. After optimisation of the initial position, the system was relaxed
without restraints. Unresolved residues and sidechains that were also built into all MD models based on the cryo-EM density. The protein was modelled in both symmetric as well as asymmetric
states, with regard to the opening angle of QcrC subunit (Supplementary Fig. 1), with a cross-correlation of 0.66 (EMDB: 9610) and 0.71 (EMDB: 0289) to respective cryo-EM maps for the
resolved parts. The LpqE subunits were removed in the asymmetric model due to steric clashes (Supplementary Fig. 1), as also indirectly supported by our biochemical assay (Fig. 6e). The MD
models were embedded in a 266 × 156 Å 1-palmitoyl-2-oleoyl-_sn_-glycero-3-phosphocholine (POPC) membrane together with eight experimentally-resolved cardiolipin residues at the dimer
interface. The MD models, comprising around 933,000–937,000 atoms, were further solvated with TIP3P water molecules and neutralised with around 783/665 Na+/Cl- ions, comparable to a 150 mM
NaCl concentration. Protein structure files were generated using CHARMM c38b84. MOLECULAR DYNAMICS SIMULATIONS Atomistic MD simulations of the III2IV2 SC model were performed using the
CHARMM36 force field for protein/lipids, ions, and water. Parameters for all cofactors were derived from in-house DFT calculations85,86, with the remaining system treated using the CHARMM36m
force field87. The MD simulations from the symmetric and asymmetric models with different menaquinone binding sites and oxidation states as well as all mutants are listed in Supplementary
Table 1. Initial protonation states were obtained from using Poisson–Boltzmann electrostatic calculations with Monte Carlo sampling88,89, based on models with the cofactors treated in their
oxidised state (see Supplementary Table 3 and Table 15, for a list of residues with non-standard protonation states and their estimated p_K_a values, respectively). All MD simulations were
performed in an _NPT_ ensemble at _T_ = 310 K and _p_ = 1 atmosphere, using a 2 fs integration time step, and with electrostatics modelled using the particle mesh Ewald (PME) method. The
system was gradually relaxed for 10 ns with harmonic restraints of 2 kcal mol−1 Å−1 on all protein and cofactor heavy atoms followed by 10 ns with harmonic restraints of 2 kcal mol−1 Å−1 on
all protein backbone heavy atoms and by 10 ns equilibration with weak (0.5 kcal mol−1 Å−1) restraints on all Cα atoms, and finally ~0.5 μs production runs. All classical MD simulations were
performed using NAMD2 (v. 2.12/2.13) for equilibration and NAMD3 (alpha9)90 for production runs. The simulations were analysed using VMD 1.9.3/1.9.491 and MDAnalysis 2.0.092,93. Hydration
dynamics and central distances were analysed based with a frequency of ca. 2 frames/ns for each trajectory. Line plots show both raw data (shaded) as well as smoothed data (solid line) using
a Savitzky-Golay filter, to improve readability. DERIVATION OF FORCE FIELD PARAMETERS Force field parameters for the redox cofactors were derived based on DFT models using a methodology
described before85,86,94,95,96,97. To this end, the DFT models were constructed based on high-resolution crystal structures and optimised at the B3LYP-D3/def2-SVP(C,H,O,N)/def2-TZVP (Cu, Fe,
S) level of theory98,99,100,101,102,103 in different oxidation and/or ligand states (Supplementary Fig. 21). Force field parameters were derived for the lowest energy spin-state
configuration, where applicable, based on the molecular Hessian104,105, and adapted together with Lennard–Jones parameters for the CHARMM36 force field87,106. Atomic partial charges were
calculated using the restrained electrostatic potential (RESP) method107,108, based on single-point calculations at the B3LYP-D3/def2-TZVP level of theory. To this end, atomic partial
charges were derived for quinone in the menaquinone (Q), menaquinol (QH2, QH−), and mena-semiquinone (QH•, Q•/−) while bonding and Lennard–Jones parameters were derived from
cgenff86,109,110. For 2His-coordinated heme _b_, and Met/His-ligated and Cys2-coordinated heme _c_ in the FeII and FeIII states both force constants and charges were derived based on DFT
models. For the Rieske centre, the DFT models were built using the 2Fe2S-2Cys-2His core, truncated at Cα atoms, and modelled as ethyl groups, and optimised without restrains. The Rieske FeS
cluster was optimised in the FeIIIFeIII and FeIIIFeII states (cf. also refs. 86,96,97,103) as well as with the imidazole group modelled in both protonated and deprotonated states
(Supplementary Fig. 21), using the broken-symmetry spin-flip approach103 with anti-ferromagnetic coupling between the metal centres. The active site of SodC was modelled with the copper, in
the oxidised state (CuII), coordinated by a hydroxyl and three His residues, which were truncated at the Cβ atom, and kept fixed during the structure optimisation. Parameters for CuA
(CuIICuI and CuICuI forms); heme _a_ (in FeIII and FeII) heme _a_3 and CuB centre (in FeIV = O2−, Cu(II)–OH− TyrO• / CuII–OH− TyrO− were adapted based on our previous work85. The redox
cofactors included surrounding first solvation sphere ligands. The charge distribution of heme _a_ and heme _a_3 thus contains the polarising effect of A/D propionates, as well as the Arg
cluster above the propionates. All DFT models were optimised using TURBOMOLE v. 7.5.1111. The force field structure and dynamics of the cofactors were compared with results from QM/MM
calculations. See Supplementary Fig. S21 for model systems used for parametrisation, and interaction validation as well as Supplementary Tables S7–S14 and ref. 85 for force field parameters.
All parameters used for the MD simulation can be found on Zenodo (repository code: https://doi.org/10.5281/zenodo.10118429) and at https://github.com/KailaLab/ff_parameters. ELECTRON
TRANSFER FLUX CALCULATIONS Electron transfer (eT) rates were estimated as described in ref. 112. Briefly, structures obtained from the MD simulations were used to obtain dynamically-averaged
eT-rates, by considering all donor-acceptor distance pairs between the cofactors for the rate calculations. For estimating approximate coupling elements113, we used the explicit intervening
protein region via calculation of a protein coupling matrix between _all_ individual eT-pathways. The approximate eT-rates were estimated based on the approximate electronic coupling
elements based on the MD simulations, experimental redox potentials, and by using generic reorganisation energies of λ = 0.7 eV, except for the heme _a_/heme _a_3 couple, where lower
reorganisation energies of λ = 0.3–0.5 eV have been proposed112,114,115. The averaged eT-rates were computed based on the individual rates (ref. 112, cf. also ref. 113), $$\log
({k}_{{ij}})=13-(1.2-0.8{\rho }_{{ij}})\left({R}_{{ij}}({{{{{{\text{\AA }}}}}}}^{-1})-3.6\right)-3.1 ({{{{{{\rm{eV}}}}}}}^{-1})\frac{{(\Delta G+\lambda )}^{2}}{\lambda }\,$$ (1)
$$\left\langle {k}_{{{{{{\rm{eT}}}}}}}\right\rangle=\,\frac{1}{N}{\sum }_{i,j}^{N}{k}_{i,j}$$ (2) The (1.2–0.8ρij)(_R_ij–3.6) factor arises from the exponential dependence of the electronic
coupling on the distance, exp(−_β_o_r_), with the _β_o ranging between 2.8 Å−1 and 3.5 Å−1 in proteins116,117, and calculated here as weighted sum of the explicit proton surroundings along
all tunnelling pathways _N_ between atom pairs _i, j_ (see also Supplementary Fig. 12). The kinetic modelling of the long-range charge transfer was simulated using COPASI 4.35118 with
microscopic rates or transition barriers estimated from the eT rate calculations, QM/MM calculation for the PCET reactions, or from experiments (see Supplementary Fig. 12, Table 4). Kinetic
models were created for both the symmetric and asymmetric complexes. The first eT from Qo1a was restricted to the Rieske FeS, while the second one was restricted from Qo1a to heme _b_L. The
eT from Qo1a to heme _b_L, and heme _a_ to heme _a_3 were modelled as irreversible to account for Qo1a, ox unbinding and oxygen reduction, respectively, resulting in a non-equilibrium state
that drives the overall reaction forward. In an alternative model, the rate of electron transfer to FeS and heme _b_L were obtained from the QM/MM calculations based on transition state
theory, leading to similar results (Supplementary Fig. 13c). A second model was used to also consider the eT within the Complex III dimer in the asymmetric system. For the open QcrC state,
with a disconnected eT chain, the Rieske FeS was assumed to be become fully reduced but blocked further eT from Qo1a along the cyt _cc_ chain. To this end, the second eT from Qo1a and the
reduction of Qi were modelled as irreversible reactions, and the electron leakage from the open to closed side of the SC was modelled via eT between heme _b_Ls cofactors of the Complex III
dimer. On the closed side of the SC, reduction of heme _a_3 was modelled as irreversible, whilst no bias was put on the eT direction in Qo1a. To account for the proton-coupled electron
transfer reaction in Complex IV, protonation on a pump site (PLS, see refs. 37,41,119) near the BNC modulated the E_m_ value of the BNC, whereas the protonation rate was modelled as the
experimentally determined 4 ms proton uptake via the D-channel, whereas the oxygen reduction was modelled as an irreversible reaction. The overall charge transfer reaction rates were
calculated from the half-time (_t_1/2) of heme _a_3 reduction using the relationship _k_ = ln 2/_t_1/2. See Data Availability for the kinetic models. WATER CLUSTER ANALYSIS Clustering of
water molecules in the MD simulations was performed using WATCLUST v0.1 plugin in VMD120 with default parameters, except for “waternumbermin”, which was set to ca. 10% of the frames, and a
“dist” value of 0.3. The water clusters were coloured by the probability of observing the water molecules at the given positions. WATER SURVIVAL PROBABILITY Water survival probabilities at
the entrance of the D-channel were calculated using the water dynamics module of MDAnalysis 2.0.092,93, selecting water molecules within 5 Å of Asp115 (bovine: D91) of subunit I in Complex
IV. See ref. 39 for further details of the simulation setup of the bovine Complex IV simulations. Survival probabilities of water molecules in the D- and K-channel were calculated using a
spherical volume with a 5 Å radius around residues Thr125 and Thr337. CHANNEL VOLUME ESTIMATION Channel volumes of the D- and K-channel were estimated for the cryo-EM structure using
CAVER3121. To this end, the channel search was initiated from Glu266/Thr182 and Tyr268/Thr337 with endpoints at Asp115 and Arg340 for the D- and K-channel, respectively. The residues at the
endpoint were excluded for the channel search and analysed using probe radii of 0.85–0.9 Å. The volume of the channel was estimated as a cylinder enclosing the region between start/endpoint
and bulk, based on the channel length and average width. REDOX POTENTIALS AND BINDING ENERGIES FROM PBE CALCULATIONS Redox potentials and p_K_a values of cofactors and amino acids were
estimated based on the continuum electrostatic calculations of the Poisson–Boltzmann equation, as implemented in APBS 1.5122 in combination with Monte Carlo sampling of protonation and redox
states, performed with Karlsberg+88,89,123. While these calculations consider thermodynamic effects, they do not account for how the substitutions affect reaction barriers or proton
pathways linked to the PCET reactions. Charges for the different oxidation and protonation states were obtained similarly as before85,86,124. The protein was described using explicit atoms
with atomic partial charges, embedded in an inhomogeneous dielectric continuum with a low dielectric constant of four. The bulk water was described by a homogeneous dielectric continuum with
a dielectric constant of 80. The boundary interface between the protein and solvent was calculated by the molecular surface routine implemented in APBS, using a solvent probe radius of 1.4
Å, and modelling an implicit ionic strength with 100 mM potassium chloride. Protonation probabilities were probed along classical simulations (Supplementary Table 1) every 1 ns. A final MC
round was performed considering only key residues and cofactors to improve convergence of the calculations. Binding energies of menaquinone in different oxidation states (QH2, QH−, QH•, Q•
and Q) were estimated at the Qo1a, Qo1b and Qo2 sites in the Complex III dimer at the continuum electrostatic level using APBS 1.5 (PBSA-MM approximation). To this end, the protein was
truncated to include subunits QcrA, QcrB, and QcrC and described with explicit point charges and a low dielectric constant of 4, and extending 5 Å beyond the protein to account for the lipid
membrane. The bulk water was described by a homogeneous dielectric continuum with a dielectric constant of 80. The boundary between protein and solvent was determined by the molecular
surface routine implemented in APBS122, using a solvent probe radius of 1.4 Å, with an implicit ionic strength with 100 mM potassium chloride. Binding energies of the quinone in the protein
were computed using a thermodynamic cycle based on contributions of desolvation effects and electrostatic interactions of quinone species and the protein environment125. All calculations
were based on the MD simulations of QH2 in Qo1a, Qo1b and Qo2 (S2, S15, S16 in Table S1) by swapping the charges of menaquinone to the desired redox state, and re-optimising the hydrogen
atom positions in Karlsberg+88,89,123 (see above). QM/MM CALCULATIONS To study the PCET reactions linked to the electron bifurcation process at Qo1a, two QM regions were defined comprising
the quinol/Rieske centre part (QM1) and the semiquinone/heme _b_L part (QM2). The two PCET reactions in QM1 and QM2 were treated as sperate models at the QM/MM level. Hybrid QM/MM umbrella
sampling (US) calculations were performed based on a structure extracted from an MD simulation of quinol obtained after 500 ns for the initial PCET step (QM1), and QM2 was constructed after
the first PCET converged to the QH• state (Fig. 2A, B). The QM1 region (195 atoms) comprised the menaquinol headgroup (up to atom C10), the FeS Rieske centre coordinated by Cys333, Cys352,
His335 and His355; and surrounding residues Leu336, Cys338, Cys354, Ser357, Phe156, Tyr159, Thr169, Ala174, Met305, Thr308, Asp309, Ile312, as well as 3 water molecules. The QM2 region (248
atoms) comprised the menaquinol (QH•) headgroup, heme _b_L with its coordinated His109 and His211, as well as Arg106, Tyr159, Asn282, Asp302, Tyr304, and eight water molecules. The MM region
comprised the Complex III dimer, and a layer of water/lipids/ions around 5 Å of the protein, resulting in a model with around 85,000 atoms. The electronic structure of the FeS Rieske centre
was prepared by anti-ferromagnetically coupling the iron atoms using the broken-symmetry spin flip approach126,127. All QM calculations were performed at the B3LYP-D398,128,100 level with
def2-SVP/def2-TZVP(Fe, S) basis sets. The QM regions were prepared with localised electrons86 at the electron donor (QH2 for the first reaction, and QH• for the reaction, see Supplementary
Fig. 11e, f). Link atoms, modelled as hydrogen atoms, were placed at the Cβ–Cα bonds, with exception of arginine residues, where the link atoms were placed between the Cγ–Cδ bond and for
menaquinone, where the link atom was placed between atom C9-C11 bond. To localise the diabatic electron transfer state, the system was relaxed separately in their corresponding redox states,
and the molecular orbitals were merged with the subsystems86. The QM/MM systems were relaxed using an adopted basis Newton-Raphson optimiser until the energy (0.0006 kcal mol−1) and
gradient convergence threshold (0.0006 kcal mol−1 Å−1) were reached. Potential energy profiles for the PCET reactions were computed based on reaction pathway optimisation for the proton
transfer reaction coordinate, constrained with a harmonic potential using a force constant of 500 kcal mol−1 Å−1, modelled as a linear combination of bond-forming and bond-breaking distances
(Supplementary Fig. 11a, b). QM/MM umbrella sampling (QM/MM-US) simulations were initiated from the intermediate states along the PCET reactions using a force constant of 100–500 kcal mol−1
Å−2 applied on the proton transfer reaction coordinate (see Supplementary Fig. 11c, d), with the QM/MM US simulations divided amongst 60 windows. The potential of mean force was computed
using the weighted histogram analysis method (WHAM) with a converge threshold set to 0.0001 kcal mol−1, and statistical errors were estimated using bootstrapping analysis. The QM/MM MD
simulations were performed at _T_ = 310 K using a 1 fs integration time step, with a 12 Å sphere around the QM region that was allowed to relax during the calculations. All QM/MM
calculations were performed using an in-house version of the CHARMM c38b/TURBOMOLE 7.5.1 python wrapper129. All DFT calculations were performed using TURBOMOLE v.7.5.1111, and the electronic
structure was visualised using VMD 1.9.3/1.9.491. To study proton donors at the Qi site, QM/MM MD simulations were performed based on the last frame from a 0.5 μs classical MD simulation
(simulation S6, Supplementary Table 1). The QM region comprised the menaquinol head group, Gln29, Lys32, Glu44, Tyr 48, Trp231, Phe257, Lys260, Ser261, 15 water molecules and one POPC giving
a total of 299 atoms and a net charge of zero. The system was minimised 15 Å around the QM region, following the reduction of the Q before starting the QM/MM MD simulations, with the MM
region comprising 78,346 atoms. Link atoms around the Qi site were placed between Cβ–Cα atoms with exception of menaquinone, with a link atom placed between the C9–C11 bond. All QM
calculations were performed at the B3LYP-D398,128,100 level with def2-SVP basis sets. The QM/MM MD simulations were performed at _T_ = 310 K using a 1 fs integration time step, and by fixing
heavy atoms around 15 Å of the QM region to avoid exchange of QM and MM water molecules. The simulations were performed for 1 ps in three replicas. To investigate possible BNC ligands in
the continuous cryo-EM density, we performed QM/MM optimisations of the BNC where the QM-regions comprised heme _a_3, His397, CuB, His264, His313, His314, Tyr268, and two water molecules
(ca. 140 atoms). In this regard, we optimised the following states: FeIII–OH−–CuII/TyrO−, FeIII–H2O–CuII/TyrO−, FeIII–OOH−–CuII/TyrO−, FeIII–OH2/−HO–CuII/TyrO−, FeIII–OH−/H2O–CuII/TyrO−,
FeIII–OH−/−HO–CuII/TyrO− and FeIII–OH-–H2O–CuII/TyrO− (see Fig. 4h, Supplementary Fig. 7e-f) at the B3LYP-D3/ def2-SVP/def2-TZVP(Cu, Fe) level of theory with the MM region composed of
remaining Complex IV system. CELL GROWTH _M. smegmatis_ mc2155 cells with the SC FLAG-tagged on QcrB, were grown in 7H9 medium, supplemented with ADS (5 g L−1 BSA, 2 g L−1 dextrose, and 0.8
g L−1 NaCl), 25 μg mL−1 kanamycin, 50 μg mL−1 hygromycin, 0.2% glycerol, and 0.05% Tween 80. Single colonies were picked from 7H9 agar plates supplemented with ADS and antibiotics,
inoculated into 25 mL culture, and shaken at 180 rpm, 30 °C. The pre-cultures were diluted after 48 h into 1000 mL 7H9 medium in a 2 L flask, and shaken at 180 rpm at 30 °C for ~72 h. Cells
were harvested after OD600 reached 2.5. MEMBRANE PREPARATION Cells were homogenised in cell lysis buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 0.5 mM EDTA) in the presence of
phenyl-methanesulfonyl fluoride and DNase I (Roche), and crushed with a cell disrupter (Constant Systems) with four cycles at 35 kPsi. Cell debris was then removed by centrifugation at
18,600 g for 30 min, followed by collection of the membranes after ultracentrifugation at 147,000 g for 90 min. ISOLATION OF THE SUPERCOMPLEX Membranes (1 g) were incubated in 10 mL of
solubilisation buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.5 mM EDTA, 2% (w/v) GDN or 1% (w/v) n-dodecyl β-d-maltoside (DDM) (Anatrace) and incubated overnight or 45 min, respectively, at
4 °C under stirring. Unsolubilised material was removed by ultracentrifugation at 147,000 g for 30 min. The supernatant was then applied to an anti-FLAG M2 column (1 mL bed volume, Sigma
Aldrich). The column was washed with four column volumes of washing buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.5 mM EDTA, 0.01% (w/v) GDN or 0.05% (w/v) DDM). Protein was eluted with 5
column volumes of elution buffer (washing buffer +5 mg/mL FLAG peptide (Sigma Aldrich)) and concentrated with a 100 kDa molecular weight cut-off concentrator (Merck Millipore). The protein
samples were aliquoted, flash-frozen in liquid N2 and stored at −80 °C. ACTIVITY ASSAYS The menaquinol oxidation: O2 reduction activity of the purified _M. smegmatis_ SC was measured by
following the O2 reduction rate upon addition of reduced substrate; 2,3-dimethyl-[1,4] naphthoquinone (Rare Chemicals GmbH) as described in refs. 22,55. The activity was measured in a buffer
solution (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.5 mM EDTA, 0.01% (w/v) GDN or 0.05% (w/v) DDM at 25 °C) using a Clark-type oxygen electrode (Hansatech instruments). The reaction was
started by addition of 5 μL of 20 mM substrate solution into an electrode chamber containing 1 mL buffered protein solution and 500 nM bovine Sod, in order to reduce the effect of the
substrate auto-oxidation (cf. ref. 12). The activity was obtained from the initial slope of the graph where the O2 concentration was linearly dependent on time. Background O2 reduction rate
in the absence of the SC was measured as a control and subtracted from that measured in the presence of the SC. The activity was calculated using the following equation:
$${Activity}\left[{e}^{-}{s}^{-1}\right]=\frac{{c}_{{ox}}\cdot 4}{60\cdot {c}_{{prot}}}$$ (3) where cox is the change in oxygen concentration over one minute, 4 is the number of electrons
needed to reduce oxygen to water and cprot is the concentration of the protein. The protein concentration was determined from difference spectra of the dithionite-reduced minus oxidised
states of the SC, recorded with a spectrophotometer (Cary 100, Agilent Technologies), and calculated using the given absorption coefficients: _ε_605–630 nm = 24 mM−1cm−1 (heme _a_),
_ε_562–577 nm = 22 mM−1cm−1 (heme _b_) and _ε_552–540 nm = 19 mM−1cm−1 (heme _c_). CRYO-EM GRID PREPARATION AND DATA COLLECTION Purified protein sample of supercomplex (3.5 mg mL−1) was
incubated with substrate (2,3-dimethyl-[1,4] naphthoquinone, 100 μM) and inhibitor (lansoprazole sulphide, Tokyo Chemical Industry, Japan; 0.5 mM) for 45 min at room temperature. After
incubation, 3 μL of sample were applied to holey carbon film coated copper EM grids (300 mesh R2/2 grid, Quantifoil, Micro Tools GmbH, Germany), blotted for 3 s at 4 °C (100% humidity) and
plunge-frozen in liquid ethane using Vitrobot Mark VI (Thermo Fisher Scientific). The grids were glow-discharged in air at 20 mA for 120 s (PELCO easiGlow) before sample application. Data
collection was performed at 300 kV using Titan Krios G3i electron microscope (Thermo Fisher Scientific) equipped with K3 Gatan detector. Dataset 1 (2.8 Å) of 18164 exposures (40 exposure
fractions each) was acquired in electron-counting mode at a nominal magnification of 105000 (0.846 Å/pixel). Automated data collection was set up using EPU software package (v2.12.1; Thermo
Fisher Scientific). Camera exposure rate was 17.6 e−/pixel/s and total exposure of the sample was 48.2 e−/Å2 (Supplementary Table 6). Dataset 2 (2.3 Å) of 24868 exposures (40 exposure
fractions each) was acquired in electron-counting mode at a nominal magnification of 105000 (0.828 Å/pixel). Automated data collection was set up using EPU software package (v2.12.1). Camera
exposure rate was 17.5 e−/pixel/s and total exposure of the sample was 40 e−/Å2 (Supplementary Table 6). DATA PROCESSING, MODEL BUILDING AND REFINEMENT The cryo-EM data was analysed using
cryoSPARC v3130 (dataset 1) and cryoSPARC v4 (dataset 2). with motion correction and CTF estimation performed using patch motion correction and CTFFIND4 (dataset 1) or patch CTF estimation
(dataset 2), respectively. For the dataset 1, templates for the particle selection were generated by manual particle picking and 2D classification, resulting in 752,267 selected particles.
The number of particle images was reduced by 2D classification to 295,370 and by further ab initio reconstruction leading to 90,918 particles. The dataset was further refined using
homogenous refinement, followed by local CTF and non-uniform refinement131 without enforcing symmetry, resulting in a final map with an overall resolution of 2.8 Å (Supplementary Fig. 16).
Templates generated for the dataset 2 were picked from over 5 million particles, with ca. 800,000 which were selected by the first round of 2D classification. Both datasets show a symmetric
supercomplex and feature both LpqE and SodC. Initial structural models were built based on previously refined structures10,11 (PDB ID: 6HWH10 and PDB ID: 6ADQ11), providing a reasonable
starting model for the refinement, followed by fitting into the map and manual adjustment using Coot 0.8.9.2132. The models contain in addition to canonical redox cofactors, ten menaquinone
molecules, 249/614 buried water molecules, and 21/38 lipid molecules (Fig. 1b, Supplementary Figs. 17). The model also contains six short additional chains on the N-side of the complex, four
of which were modelled as poly-alanine residues due to unresolved sidechains. Model refinement was performed using real-space refinement in Phenix 1.118.2133 and Refmac 5.8.0257 from
CCP-EM134 as well as MD flexible fitting as implemented in NAMD2.13, with manual adjustments in Coot. The structural features of the model were validated using MolProbity135. Examples
densities, rendered in UCSF ChimeraX 1.5136, are shown in Figs. 1,3,4 and Supplementary Figs. 7, 10, 17, with map thresholds given based on the map level in UCSF ChimeraX. The refined
structure and density are deposited with access codes: PDB ID: 8OVD and 8OVC and EMBD: 17211 and 17,210 for the 2.3 Å and 2.8 Å maps, respectively. REPORTING SUMMARY Further information on
research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The data that support this study are available from the corresponding authors
upon request. Cryo-EM maps are available in the Electron Microscopy Data Bank (EMD–17211 [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-17211] (III2IV2-SC, 2.3 Å map) and EMD-17210 (III2IV2-SC,
2.8 Å map)), and atomic models of the _M. smegmatis_ supercomplex are available in the Protein Data Bank (PDB – 8OVD (III2IV2-SC, 2.3 Å model) and 8OVC (III2IV2-SC, 2.8 Å model)). Previous
structures, referred to in the manuscript, can found on the Protein Data Bank with the following accession codes: 6HWH (III2IV2-SC), 6ADQ (III2IV2-SC), 7RH5 (III2IV2-SC), 7RH6 (III2IV2-SC),
7RH7 (III2IV2-SC), 7E1V (III2IV2-SC), 7QHO (III2IV2-SC), 7COH (Complex IV), 7ATE (Complex IV), 7AU6 (Complex IV), 6Q9E (Complex III), 1PP9 (Complex III), 1KYO (Complex III), 1PZS (SOD).
Cryo-EM maps from previous structures used to calculate cross-correlation can be access in the Electron Microscopy Data Bank (EMD–0289 [https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-0289]
(III2IV2-SC, 6HWH) and EMD-9610 (III2IV2-SC, 6ADQ)). Models, parameters, and key trajectories have been deposited to the Zenodo database [https://doi.org/10.5281/zenodo.10118429]. REFERENCES
* Kaila, V. R. I. & Wikström, M. Architecture of bacterial respiratory chains. _Nat. Rev. Microbiol._ 19, 319–330 (2021). Article CAS PubMed Google Scholar * Mitchell, P. Coupling
of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. _Nature_ 191, 144–148 (1961). Article ADS CAS PubMed Google Scholar * Schägger, H. &
Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. _EMBO J._ 19, 1777–1783 (2000). Article PubMed PubMed Central Google Scholar * Davies, K. M.,
Blum, T. B. & Kühlbrandt, W. Conserved in situ arrangement of complex I and III2 in mitochondrial respiratory chain supercomplexes of mammals, yeast, and plants. _Proc. Nat. Acad. Sci.
USA_ 115, 3024–3029 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Brzezinski, P., Moe, A. & Ädelroth, P. Structure and mechanism of respiratory III–IV
supercomplexes in bioenergetic membranes. _Chem. Rev._ 121, 9644–9673 (2021). * Trouillard, M., Meunier, B. & Rappaport, F. Questioning the functional relevance of mitochondrial
supercomplexes by time-resolved analysis of the respiratory chain. _Proc. Nat. Acad. Sci. USA_ 108, E1027–E1034 (2011). Article ADS CAS PubMed PubMed Central Google Scholar *
Milenkovic, D., Blaza, J. N., Larsson, N.-G. & Hirst, J. The enigma of the respiratory chain supercomplex. _Cell Metab._ 25, 765–776 (2017). Article CAS PubMed Google Scholar *
Fedor, J. G. & Hirst, J. Mitochondrial supercomplexes do not enhance catalysis by quinone channeling. _Cell Metab._ 28, 525–531.e4 (2018). Article CAS PubMed PubMed Central Google
Scholar * Stuchebrukhov, A., Schäfer, J., Berg, J. & Brzezinski, P. Kinetic advantage of forming respiratory supercomplexes. _Biochim. Biophys. Acta Bioenerg._ 1861, 148193 (2020).
Article CAS PubMed Google Scholar * Wiseman, B. et al. Structure of a functional obligate complex III2IV2 respiratory supercomplex from Mycobacterium smegmatis. _Nat. Struct. Mol. Biol._
25, 1128–1136 (2018). Article CAS PubMed Google Scholar * Gong, H. et al. An electron transfer path connects subunits of a mycobacterial respiratory supercomplex. _Science_ 362,
eaat8923 (2018). Article ADS PubMed Google Scholar * Yanofsky, D. J. et al. Structure of mycobacterial CIII2CIV2 respiratory supercomplex bound to the tuberculosis drug candidate
telacebec (Q203). _eLife_ 10, e71959 (2021). Article CAS PubMed PubMed Central Google Scholar * Zhou, S. et al. Structure of Mycobacterium tuberculosis cytochrome _bcc_ in complex with
Q203 and TB47, two anti-TB drug candidates. _eLife_ 10, e69418 (2021). Article CAS PubMed PubMed Central Google Scholar * Liang, Y. & Rubinstein, J. L. Structural analysis of
mycobacterial electron transport chain complexes by cryoEM. _Biochem. Soc. Trans._ 51, 183–193 (2023). CAS PubMed Google Scholar * Kao, W.-C. et al. Structural basis for safe and
efficient energy conversion in a respiratory supercomplex. _Nat. Commun._ 13, 545 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Letts, J. A. et al. Structures of
respiratory supercomplex I + III2 reveal functional and conformational crosstalk. _Mol. Cell_ 75, 1131–1146.e6 (2019). Article CAS PubMed PubMed Central Google Scholar * Hartley, A. M.
et al. Structure of yeast cytochrome c oxidase in a supercomplex with cytochrome bc1. _Nat. Struct. Mol. Biol._ 26, 78–83 (2019). Article CAS PubMed Google Scholar * Letts, J. A.,
Fiedorczuk, K. & Sazanov, L. A. The architecture of respiratory supercomplexes. _Nature_ 537, 644–648 (2016). Article ADS CAS PubMed Google Scholar * Gu, J. et al. The architecture
of the mammalian respirasome. _Nature_ 537, 639–643 (2016). Article ADS CAS PubMed Google Scholar * Guo, R. et al. Architecture of human mitochondrial respiratory megacomplex I2III2IV2.
_Cell_ 170, 1247–1257.e12 (2017). Article CAS PubMed Google Scholar * Mühleip, A. et al. Structural basis of mitochondrial membrane bending by the I–II–III2–IV2 supercomplex. _Nature_
615, 934–938 (2023). Article ADS PubMed PubMed Central Google Scholar * Moe, A. et al. The respiratory supercomplex from _C. glutamicum_. _Structure_ 30, 338–349.e3 (2022). Article CAS
PubMed Google Scholar * WHO. Global Tuberculosis reports. https://www.who.int/teams/global-tuberculosis-programme/tb-reports (2021). * Kaila, V. R. I., Verkhovsky, M. I. & Wikström,
M. Proton-coupled electron transfer in cytochrome _c_ oxidase. _Chem. Rev._ 110, 7062–7081 (2010). Article CAS PubMed Google Scholar * Wikström, M. et al. New perspectives on proton
pumping in cellular respiration. _Chem. Rev._ 115, 2196–2221 (2015). Article PubMed Google Scholar * Crofts, A. R. The modified Q-cycle: a look back at its development and forward to a
functional model. _Biochim. Biophys. Acta Bioenerg._ 1862, 148417 (2021). Article CAS PubMed Google Scholar * Mitchell, P. The protonmotive Q cycle: a general formulation. _FEBS Lett._
59, 137–139 (1975). Article CAS PubMed Google Scholar * Mitchell, P. Possible molecular mechanisms of the protonmotive function of cytochrome systems. _J. Theor. Biol._ 62, 327–367
(1976). Article ADS CAS PubMed Google Scholar * Crofts, A. R. et al. Pathways for proton release during ubihydroquinone oxidation by the _bc_1 complex. _Proc. Nat. Acad. Sci. USA_ 96,
10021–10026 (1999). Article ADS CAS PubMed PubMed Central Google Scholar * Postila, P. A. et al. Atomistic determinants of co-enzyme Q reduction at the Qi-site of the cytochrome bc1
complex. _Sci. Rep._ 6, 33607 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Crofts, A. R., Hong, S., Zhang, Z. & Berry, E. A. Physicochemical aspects of the
movement of the Rieske iron sulfur protein during quinol oxidation by the _bc_1 complex from mitochondria and photosynthetic bacteria. _Biochemistry_ 38, 15827–15839 (1999). Article CAS
PubMed Google Scholar * Sarewicz, M. et al. Catalytic reactions and energy conservation in the cytochrome _bc_1 and _b_6_f_ complexes of energy-transducing membranes. _Chem. Rev._ 121,
2020–2108 (2021). Article CAS PubMed PubMed Central Google Scholar * Crofts, A. R. Proton-coupled electron transfer at the Qo-site of the bc1 complex controls the rate of
ubihydroquinone oxidation. _Biochim. Biophys. Acta Bioenerg._ 1655, 77–92 (2004). Article CAS Google Scholar * Crofts, A. R. et al. The mechanism of ubihydroquinone oxidation at the
Qo-site of the cytochrome bc1 complex. _Biochim. Biophys. Acta Bioenerg._ 1827, 1362–1377 (2013). Article CAS Google Scholar * Wilson, C. A. & Crofts, A. R. Dissecting the pattern of
proton release from partial process involved in ubihydroquinone oxidation in the Q-cycle. _Biochim. Biophys. Acta Bioenerg._ 1859, 531–543 (2018). Article CAS PubMed Google Scholar *
Barragan, A. M. et al. Theoretical description of the primary proton-coupled electron transfer reaction in the cytochrome bc1 complex. _J. Am. Chem. Soc._ 143, 715–723 (2021). Article CAS
PubMed PubMed Central Google Scholar * Belevich, I., Verkhovsky, M. I. & Wikström, M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. _Nature_ 440,
829–832 (2006). Article ADS CAS PubMed Google Scholar * Supekar, S. & Kaila, V. R. I. Dewetting transitions coupled to K-channel activation in cytochrome _c_ oxidase. _Chem. Sci._
9, 6703–6710 (2018). Article CAS PubMed PubMed Central Google Scholar * Di Trani, J. M. et al. Structural basis of mammalian complex IV inhibition by steroids. _Proc. Nat. Acad. Sci.
USA_ 119, e2205228119 (2022). Article CAS PubMed PubMed Central Google Scholar * Wikström, M., Verkhovsky, M. I. & Hummer, G. Water-gated mechanism of proton translocation by
cytochrome c oxidase. _Biochim. Biophys. Acta Bioenerg._ 1604, 61–65 (2003). Article Google Scholar * Saura, P. et al. Electric fields control water-gated proton transfer in cytochrome _c_
oxidase. _Proc. Nat. Acad. Sci. USA_ 119, e2207761119 (2022). Article CAS PubMed PubMed Central Google Scholar * Kaila, V. R. I., Verkhovsky, M. I., Hummer, G. & Wikström, M.
Glutamic acid 242 is a valve in the proton pump of cytochrome _c_ oxidase. _Proc. Nat. Acad. Sci. USA_ 105, 6255–6259 (2008). Article ADS CAS PubMed PubMed Central Google Scholar * Lu,
J. & Gunner, M. R. Characterizing the proton loading site in cytochrome c oxidase. _Proc. Nat. Acad. Sci. USA_ 111, 12414–12419 (2014). Article ADS CAS PubMed PubMed Central Google
Scholar * Namslauer, A., Aagaard, A., Katsonouri, A. & Brzezinski, P. Intramolecular proton-transfer reactions in a membrane-bound proton pump: the effect of pH on the peroxy to ferryl
transition in cytochrome _c_ oxidase. _Biochemistry_ 42, 1488–1498 (2003). Article CAS PubMed Google Scholar * Kaila, V. R. I., Sharma, V. & Wikström, M. The identity of the
transient proton loading site of the proton-pumping mechanism of cytochrome _c_ oxidase. _Biochim. Biophys. Acta_ 1807, 80–84 (2011). Article CAS PubMed Google Scholar * Kao, W.-C. &
Hunte, C. Quinone binding sites of cyt bc complexes analysed by X-ray crystallography and cryogenic electron microscopy. _Biochem. Soc. Trans._ 50, 877–893 (2022). Article CAS PubMed
PubMed Central Google Scholar * Osyczka, A. et al. Role of the PEWY glutamate in hydroquinone−quinone oxidation−reduction catalysis in the Qo site of cytochrome bc1. _Biochemistry_ 45,
10492–10503 (2006). Article CAS PubMed Google Scholar * Kao, W.-C. & Hunte, C. The molecular evolution of the Qo motif. _Genome Biol. Evol._ 6, 1894–1910 (2014). Article PubMed
PubMed Central Google Scholar * Kao, W.-C. et al. The obligate respiratory supercomplex from Actinobacteria. _Biochim. Biophys. Acta Bioenerg._ 1857, 1705–1714 (2016). Article CAS Google
Scholar * Zu, Y. et al. Reduction potentials of Rieske clusters: importance of the coupling between oxidation state and histidine protonation state. _Biochemistry_ 42, 12400–12408 (2003).
Article CAS PubMed Google Scholar * Brandt, U. Bifurcated ubihydroquinone oxidation in the cytochrome _bc_1 complex by proton‐gated charge transfer. _FEBS Lett._ 387, 1–6 (1996).
Article ADS CAS PubMed Google Scholar * Zhang, P. et al. Electron bifurcation: thermodynamics and kinetics of two-electron brokering in biological redox chemistry. _Acc. Chem. Res._ 50,
2410–2417 (2017). Article CAS PubMed Google Scholar * Müller, V., Chowdhury, N. P. & Basen, M. Electron bifurcation: a long-hidden energy-coupling mechanism. _Annu. Rev. Microbiol._
72, 331–353 (2018). Article PubMed Google Scholar * Yuly, J. L. et al. Electron bifurcation: progress and grand challenges. _Chem. Commun._ 55, 11823–11832 (2019). Article CAS Google
Scholar * Król, S. et al. Electron and proton transfer in the M. smegmatis III2IV2 supercomplex. _Biochim. Biophys. Acta Bioenerg._ 1863, 148585 (2022). Article PubMed Google Scholar *
Graf, S. et al. Rapid electron transfer within the III-IV supercomplex in Corynebacterium glutamicum. _Sci. Rep._ 6, 34098 (2016). Article ADS CAS PubMed PubMed Central Google Scholar
* Ädelroth, P., Ek, M. & Brzezinski, P. Factors determining electron-transfer rates in cytochrome _c_ oxidase: investigation of the oxygen reaction in the _R. sphaeroides_ enzyme.
_Biochim Biophys. Acta - Bioenerg._ 1367, 107–117 (1998). Article Google Scholar * Namslauer, A. & Brzezinski, P. Structural elements involved in electron-coupled proton transfer in
cytochrome _c_ oxidase. _FEBS Lett._ 567, 103–110 (2004). CAS PubMed Google Scholar * Kleinschroth, T. et al. X-ray structure of the dimeric cytochrome _bc_1 complex from the soil
bacterium Paracoccus denitrificans at 2.7-Å resolution. _Biochim. Biophys. Acta Bioenerg._ 1807, 1606–1615 (2011). Article CAS Google Scholar * Czapla, M., Borek, A., Sarewicz, M. &
Osyczka, A. Enzymatic activities of isolated cytochrome _bc_1-like complexes containing fused cytochrome _b_ subunits with asymmetrically inactivated segments of electron transfer chains.
_Biochemistry_ 51, 829–835 (2012). Article CAS PubMed Google Scholar * Castellani, M. et al. Direct demonstration of Half-of-the-sites reactivity in the dimeric cytochrome _bc_1 complex.
_J. Biol. Chem._ 285, 502–510 (2010). Article CAS PubMed Google Scholar * Świerczek, M. et al. An electronic bus bar lies in the core of cytochrome _bc_1. _Science_ 329, 451–454 (2010).
Article ADS PubMed PubMed Central Google Scholar * Lange, C., Nett, J. H., Trumpower, B. L. & Hunte, C. Specific roles of protein-phospholipid interactions in the yeast cytochrome
_bc_1 complex structure. _EMBO J._ 20, 6591–6600 (2001). Article CAS PubMed PubMed Central Google Scholar * Shinzawa-Itoh, K. et al. The 1.3-Å resolution structure of bovine cytochrome
c oxidase suggests a dimerization mechanism. _BBA Adv._ 1, 100009 (2021). Article CAS PubMed PubMed Central Google Scholar * Kolbe, F. et al. Cryo-EM structures of intermediates suggest
an alternative catalytic reaction cycle for cytochrome c oxidase. _Nat. Commun._ 12, 6903 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Tsukihara, T. et al. The whole
structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. _Science_ 272, 1136–1144 (1996). Article ADS CAS PubMed Google Scholar * Rich, P. R. & Maréchal, A. Functions of
the hydrophilic channels in protonmotive cytochrome _c_ oxidase. _J. R. Soc. Interface_ 10, 20130183 (2013). Article PubMed PubMed Central Google Scholar * Kaila, V. R. et al. A
combined quantum chemical and crystallographic study on the oxidized binuclear center of cytochrome c oxidase. _Biochim. Biophys. Acta_ 1807, 769–778 (2011). Article CAS PubMed Google
Scholar * Wikström, M., Gennis, R. B. & Rich, P. R. Structures of the intermediates in the catalytic cycle of mitochondrial cytochrome _c_ oxidase. _Biochim. Biophys. Acta Bioenerg._
1864, 148933 (2023). Article PubMed Google Scholar * Ostermeier, C., Harrenga, A., Ermler, U. & Michel, H. Structure at 2.7 Å resolution of the _Paracoccus denitrificans_ two-subunit
cytochrome _c_ oxidase complexed with an antibody FV fragment. _Proc. Nat. Acad. Sci. USA_ 94, 10547–10553 (1997). Article ADS CAS PubMed PubMed Central Google Scholar * Maréchal, A.,
Iwaki, M. & Rich, P. R. Structural changes in cytochrome c oxidase induced by binding of sodium and calcium ions: An ATR-FTIR study. _J. Am. Chem. Soc._ 135, 5802–5807 (2013). Article
PubMed Google Scholar * Vygodina, T., Kirichenko, A. & Konstantinov, A. A. Direct regulation of cytochrome _c_ Oxidase by calcium ions. _PLoS ONE_ 8, e74436 (2013). Article ADS CAS
PubMed PubMed Central Google Scholar * Berndtsson, J. et al. Respiratory supercomplexes enhance electron transport by decreasing cytochrome _c_ diffusion distance. _EMBO Rep._ 21, e51015
(2020). Article CAS PubMed PubMed Central Google Scholar * Bansal-Mutalik, R. & Nikaido, H. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich
in diacyl phosphatidylinositol dimannosides. _Proc. Nat. Acad. Sci. USA_ 111, 4958–4963 (2014). Article ADS CAS PubMed PubMed Central Google Scholar * Zhang, Z. et al. Electron
transfer by domain movement in cytochrome. _bc1. Nat._ 392, 677–684 (1998). Article CAS Google Scholar * Piddington, D. L. et al. Cu, Zn superoxide dismutase of _Mycobacterium
tuberculosis_ contributes to survival in activated macrophages that are generating an oxidative burst. _Infect. Immun._ 69, 4980–4987 (2001). Article CAS PubMed PubMed Central Google
Scholar * Liao, D., Fan, Q. & Bao, L. The role of superoxide dismutase in the survival of _Mycobacterium tuberculosis_ in macrophages. _Jpn. J. Infect. Dis._ 66, 480–488 (2013). Article
PubMed Google Scholar * Spagnolo, L. et al. Unique features of the sodC-encoded superoxide dismutase from Mycobacterium tuberculosis, a fully functional copper-containing enzyme lacking
zinc in the active site. _J. Biol. Chem._ 279, 33447–33455 (2004). Article CAS PubMed Google Scholar * Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and
complexes. _Nucleic Acids Res._ 46, W296–W303 (2018). Article CAS PubMed PubMed Central Google Scholar * Sakamoto, J. et al. Cytochrome c oxidase contains an extra charged amino acid
cluster in a new type of respiratory chain in the amino-acid-producing Gram-positive bacterium Corynebacterium glutamicum. The GenBank/EMBL/DDBJ accession numbers for the sequences reported
in this. _Microbiology_ 147, 2865–2871 (2001). Article CAS PubMed Google Scholar * Noguchi, S. et al. Over-expression of membrane-bound cytochrome c-551 from thermophilic Bacillus PS3 in
Bacillus stearothermophilus K1041. _Biochim Biophys. Acta - Bioenerg._ 1188, 302–310 (1994). Article Google Scholar * Birth, D., Kao, W.-C. & Hunte, C. Structural analysis of
atovaquone-inhibited cytochrome _bc_1 complex reveals the molecular basis of antimalarial drug action. _Nat. Commun._ 5, 4029 (2014). Article ADS CAS PubMed Google Scholar * Huang,
L.-S., Cobessi, D., Tung, E. Y. & Berry, E. A. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered
intramolecular hydrogen-bonding pattern. _J. Mol. Biol._ 351, 573–597 (2005). Article CAS PubMed PubMed Central Google Scholar * Brooks, B. R. et al. CHARMM: the biomolecular simulation
program. _J. Comput. Chem._ 30, 1545–1614 (2009). Article CAS PubMed PubMed Central Google Scholar * Johansson, M. P., Kaila, V. R. I. & Laakkonen, L. Charge parameterization of
the metal centers in cytochrome _c_ oxidase. _J. Comput. Chem._ 29, 753–767 (2008). Article CAS PubMed Google Scholar * Gamiz-Hernandez, A. P., Jussupow, A., Johansson, M. P. &
Kaila, V. R. I. Terminal electron-proton transfer dynamics in the quinone reduction of respiratory complex I. _J. Am. Chem. Soc._ 139, 16282–16288 (2017). Article CAS PubMed PubMed
Central Google Scholar * Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. _Nat. Methods_ 14, 71–73 (2017). Article CAS PubMed Google
Scholar * Kieseritzky, G. & Knapp, E. W. Optimizing p_K_a computation in proteins with pH adapted conformations. _Proteins_ 71, 1335–1348 (2008). Article CAS PubMed Google Scholar
* Kieseritzky, G. & Knapp, E. W. Improved p_K_a prediction: combining empirical and semimicroscopic methods. _J. Comput. Chem._ 29, 2575–2581 (2008). Article CAS PubMed Google Scholar
* Phillips, J. C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. _J. Chem. Phys._ 153, 044130 (2020). Article CAS PubMed PubMed Central Google Scholar *
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. _J. Mol. Graph._ 14, 33–38 (1996). Article CAS PubMed Google Scholar * Michaud-Agrawal, N., Denning, E. J.,
Woolf, T. B. & Beckstein, O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. _J. Comput. Chem._ 32, 2319–2327 (2011). Article CAS PubMed PubMed Central
Google Scholar * Gowers, R. et al. MDAnalysis: A python package for the rapid analysis of molecular dynamics simulations. In _Proc. of the Python in Science Conference_ (SciPy, 2016). *
Smith, D. M. et al. Force-field development and molecular dynamics of [NiFe] hydrogenase. _J. Chem. Theory Comput_ 8, 2103–2114 (2012). Article CAS PubMed Google Scholar * Di Luca, A.,
Gamiz-Hernandez, A. P. & Kaila, V. R. I. Symmetry-related proton transfer pathways in respiratory complex I. _Proc. Natl Acad. Sci. USA_ 114, E6314–E6321 (2017). PubMed PubMed Central
Google Scholar * Muhlbauer, M. E., Gamiz-Hernandez, A. P. & Kaila, V. R. I. Functional dynamics of an ancient membrane-bound hydrogenase. _J. Am. Chem. Soc._ 143, 20873–20883 (2021).
Article CAS PubMed PubMed Central Google Scholar * Katsyv, A. et al. Molecular basis of the electron bifurcation mechanism in the [FeFe]-hydrogenase complex HydABC. _J. Am. Chem. Soc_.
(in the press) (2023). * Lee, C., Yang, W. & Parr, G. P. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. _Phys. Rev. B_ 37,
785–789 (1988). Article ADS CAS Google Scholar * Schäfer, A., Horn, H. & Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. _J. Chem. Phys._ 97,
2571–2577 (1988). Article ADS Google Scholar * Becke, A. D. Density‐functional thermochemistry. III. The role of exact exchange. _J. Chem. Phys_. 98, 5648–5652 (1993). * Grimme, S.,
Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. _J. Chem. Phys._
132, 154104 (2010). Article ADS PubMed Google Scholar * Klamt, A. & Schüürmann, G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the
screening energy and its gradient. _J. Chem. Soc. Perkin Trans_. 2, 799–805 (1993). * Noodleman, L. & Davidson, E. R. Ligand spin polarization and antiferromagnetic coupling in
transition metal dimers. _Chem. Phys._ 109, 131–143 (1986). Article Google Scholar * Nilsson, K., Lecerof, D., Sigfridsson, E. & Ryde, U. An automatic method to generate force-field
parameters for hetero-compounds. _Acta Crystallogr. D Biol. Crystallogr._ 59, 274–289 (2003). Article ADS PubMed Google Scholar * Allen, A. E. A., Payne, M. C. & Cole, D. J. Harmonic
force constants for molecular mechanics force fields via Hessian matrix projection. _J. Chem. Theory Comput._ 14, 274–281 (2018). Article CAS PubMed Google Scholar * Jo, S., Kim, T.,
Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. _J. Comput. Chem._ 29, 1859–1865 (2008). Article CAS PubMed Google Scholar * Woods, R. J. &
Chappelle, R. Restrained electrostatic potential atomic partial charges for condensed-phase simulations of carbohydrates. _J. Mol. Struct._ 527, 149–156 (2000). Article CAS Google Scholar
* Cornell, W. D., Cieplak, P., Bayly, C. I. & Kollman, P. A. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation.
_J. Am. Chem. Soc._ 115, 9620–9631 (1993). Article CAS Google Scholar * Vanommeslaeghe, K. & MacKerell, A. D. Jr. Automation of the CHARMM general force field (CGenFF) I: bond
perception and atom typing. _J. Chem. Inf. Model._ 52, 3144–3154 (2012). Article CAS PubMed PubMed Central Google Scholar * Vanommeslaeghe, K., Raman, E. P. & MacKerell, A. D. Jr.
Automation of the CHARMM general force field (CGenFF) II: assignment of bonded parameters and partial atomic charges. _J. Chem. Inf. Model._ 52, 3155–3168 (2012). Article CAS PubMed
PubMed Central Google Scholar * Balasubramani, S. G. et al. TURBOMOLE: modular program suite for ab initio quantum-chemical and condensed-matter simulations. _J. Chem. Phys._ 152, 184107
(2020). Article ADS CAS PubMed PubMed Central Google Scholar * Kaila, V. R. I., Johansson, M. P., Sundholm, D. & Wikström, M. Interheme electron tunneling in cytochrome _c_
oxidase. _Proc. Nat. Acad. Sci. USA_ 107, 21470–21475 (2010). Article ADS CAS PubMed PubMed Central Google Scholar * Page, C. C., Moser, C. C., Chen, X. & Dutton, P. L. Natural
engineering principles of electron tunnelling in biological oxidation–reduction. _Nature_ 402, 47–52 (1999). Article ADS CAS PubMed Google Scholar * Jasaitis, A. et al. Activationless
electron transfer through the hydrophobic core of cytochrome _c_ oxidase. _Proc. Nat. Acad. Sci. Usa._ 102, 10882–10886 (2005). Article ADS CAS PubMed PubMed Central Google Scholar *
Blumberger, J. Recent advances in the theory and molecular simulation of biological electron transfer reactions. _Chem. Rev._ 115, 11191–11238 (2015). Article CAS PubMed Google Scholar *
Beratan, D. N., Onuchic, J. N., Winkler, J. R. & Gray, H. B. Electron-tunneling pathways in proteins. _Science_ 258, 1740–1741 (1992). Article ADS CAS PubMed Google Scholar *
Moser, C. C. et al. Nature of biological electron transfer. _Nature_ 355, 796–802 (1992). Article ADS CAS PubMed Google Scholar * Hoops, S. et al. COPASI—a COmplex PAthway SImulator.
_Bioinformatics_ 22, 3067–3074 (2006). Article CAS PubMed Google Scholar * Belevich, I. et al. Exploring the proton pump mechanism of cytochrome _c_ oxidase in real time. _Proc. Nat.
Acad. Sci. USA_ 104, 2685–2690 (2007). Article ADS CAS PubMed PubMed Central Google Scholar * López, E. D. et al. WATCLUST: a tool for improving the design of drugs based on
protein-water interactions. _Bioinformatics_ 31, 3697–3699 (2015). Article PubMed Google Scholar * Chovancova, E. et al. CAVER 3.0: a tool for the analysis of transport pathways in
dynamic protein structures. _PLoS Comput. Biol._ 8, e1002708 (2012). Article CAS PubMed PubMed Central Google Scholar * Baker, N. A. et al. Electrostatics of nanosystems: application to
microtubules and the ribosome. _Proc. Nat. Acad. Sci. USA_ 98, 10037–10041 (2001). Article ADS CAS PubMed PubMed Central Google Scholar * Gámiz-Hernández, A. P., Galstyan, A. S. &
Knapp, E.-W. Understanding rubredoxin redox potentials: role of H-bonds on model complexes. _J. Chem. Theory Comput._ 5, 2898–2908 (2009). Article PubMed Google Scholar *
Gámiz-Hernández, A. P. et al. Understanding properties of cofactors in proteins: redox potentials of synthetic cytochromes b. _ChemPhysChem_ 11, 1196–1206 (2010). Article PubMed Google
Scholar * Chakavorty, A., Li, L. & Alexov, E. Electrostatic component of binding energy: Interpreting predictions from Poisson-Boltzmann equation and modeling protocols. _J. Comput.
Chem._ 37, 2495–2507 (2016). Article CAS PubMed PubMed Central Google Scholar * Noodleman, L. Valence bond description of antiferromagnetic coupling in transition metal dimers. _J.
Chem. Phys._ 74, 5737–5743 (1981). Article ADS CAS Google Scholar * Yamaguchi, K. et al. Development of broken-symmetry (BS) methods in chemical reactions. A theoretical view of water
oxidation in photosystem II and related systems. _J. Photochem. Photobiol. A Chem._ 402, 112791 (2020). Article CAS Google Scholar * Schäfer, A., Huber, C., & Ahlrichs, R. Fully
Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. _J. Chem. Phys._ 100, 5829 (1994). Article ADS Google Scholar * Riahi, S. & Rowley, C. N.
The CHARMM-TURBOMOLE interface for efficient and accurate QM/MM molecular dynamics, free energies, and excited state properties. _J. Comput. Chem._ 35, 2076–2086 (2014). Article CAS PubMed
Google Scholar * Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. _Nat. Methods_ 14,
290–296 (2017). Article CAS PubMed Google Scholar * Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM
reconstruction. _Nat. Methods_ 17, 1214–1221 (2020). Article CAS PubMed Google Scholar * Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of _Coot_. _Acta
Crystallogr. D. Biol. Crystallogr._ 66, 486–501 (2010). Article ADS CAS PubMed PubMed Central Google Scholar * Afonine, P. V. et al. Real-space refinement in _PHENIX_ for cryo-EM and
crystallography. _Acta Crystallogr. D. Biol. Crystallogr._ 74, 531–544 (2018). Article ADS CAS Google Scholar * Brown, A. et al. Tools for macromolecular model building and refinement
into electron cryo-microscopy reconstructions. _Acta Crystallogr. D. Biol. Crystallogr._ 71, 136–153 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Chen, V. B. et al.
_MolProbity_: all-atom structure validation for macromolecular crystallography. _Acta Crystallogr. D. Biol. Crystallogr._ 66, 12–21 (2010). Article ADS CAS PubMed Google Scholar *
Pettersen, E. F. et al. UCSF Chimera-A visualization system for exploratory research and analysis. _J. Comput. Chem._ 25, 1605–1612 (2004). Article CAS PubMed Google Scholar Download
references ACKNOWLEDGEMENTS The project was supported by the Knut and Alice Wallenberg Foundation (KAW 2019.0043 to VRIK, MH, and PB, and KAW 2019.0251 to VRIK) and the Swedish Research
Council. This work was also supported by the Swedish National Infrastructure for Computing (SNIC/NAISS 2022/1-29, 2021/1-40, 2020/1-38 to VRIK) at Centre for High Performance Computing (PDC)
Centre, partially funded by the Swedish Research Council through Grant Agreement 016-07213, as well as LUMI-Sweden (project: 2022/13-14 to VRIK), and the Leibniz Rechenzentrum (LRZ,
project:pr83ro to VRIK). The cryo-EM data were collected at the Cryo-EM Swedish National Facility funded by the Knut and Alice Wallenberg, Family Erling Persson and Kempe Foundations,
SciLifeLab, Stockholm University and Umeå University. FUNDING Open access funding provided by Stockholm University. AUTHOR INFORMATION Author notes * These authors contributed equally:
Daniel Riepl, Ana P. Gamiz-Hernandez, Terezia Kovalova. AUTHORS AND AFFILIATIONS * Department of Biochemistry and Biophysics, The Arrhenius Laboratories for Natural Sciences, Stockholm
University, SE-106 91, Stockholm, Sweden Daniel Riepl, Ana P. Gamiz-Hernandez, Terezia Kovalova, Sylwia M. Król, Sophie L. Mader, Dan Sjöstrand, Martin Högbom, Peter Brzezinski & Ville
R. I. Kaila Authors * Daniel Riepl View author publications You can also search for this author inPubMed Google Scholar * Ana P. Gamiz-Hernandez View author publications You can also search
for this author inPubMed Google Scholar * Terezia Kovalova View author publications You can also search for this author inPubMed Google Scholar * Sylwia M. Król View author publications You
can also search for this author inPubMed Google Scholar * Sophie L. Mader View author publications You can also search for this author inPubMed Google Scholar * Dan Sjöstrand View author
publications You can also search for this author inPubMed Google Scholar * Martin Högbom View author publications You can also search for this author inPubMed Google Scholar * Peter
Brzezinski View author publications You can also search for this author inPubMed Google Scholar * Ville R. I. Kaila View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS V.R.I.K. designed the study; D.R., A.P.G.H., S.L.M., V.R.I.K performed molecular simulations; T.K. prepared cryo-EM grids, collected cryo-EM data, processed
cryo-EM data; T.K., D.R., A.P.G.H., V.R.I.K. built cryo-EM models; T.K., D.R., A.P.G.H., V.R.I.K. analysed and interpreted the cryo-EM models; S.M.K. isolated and biochemically characterised
the protein; D.R., A.P.G.H., T.K., S.M.K., S.L.M., D.S., M.H., P.B., V.R.I.K. analysed the data; D.R., A.P.G.H. developed new analytical tools; V.R.I.K. directed the project; V.R.I.K., D.R.
and A.P.G.H. wrote the manuscript with contribution from all authors. CORRESPONDING AUTHOR Correspondence to Ville R. I. Kaila. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare
no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file
is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY MOVIE 1 SUPPLEMENTARY MOVIE 2 SUPPLEMENTARY MOVIE 3 SUPPLEMENTARY MOVIE 4
SUPPLEMENTARY MOVIE 5 REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use,
sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative
Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Riepl, D., Gamiz-Hernandez, A.P., Kovalova, T. _et al._ Long-range charge transfer mechanism of the III2IV2 mycobacterial supercomplex. _Nat
Commun_ 15, 5276 (2024). https://doi.org/10.1038/s41467-024-49628-9 Download citation * Received: 16 November 2023 * Accepted: 12 June 2024 * Published: 20 June 2024 * DOI:
https://doi.org/10.1038/s41467-024-49628-9 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative