Structural basis for the modulation of MRP2 activity by phosphorylation and drugs
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Multidrug resistance-associated protein 2 (MRP2/ABCC2) is a polyspecific efflux transporter of organic anions expressed in hepatocyte canalicular membranes. MRP2 dysfunction, in
Dubin-Johnson syndrome or by off-target inhibition, for example by the uricosuric drug probenecid, elevates circulating bilirubin glucuronide and is a cause of jaundice. Here, we determine
the cryo-EM structure of rat Mrp2 (rMrp2) in an autoinhibited state and in complex with probenecid. The autoinhibited state exhibits an unusual conformation for this class of transporter in
which the regulatory domain is folded within the transmembrane domain cavity. In vitro phosphorylation, mass spectrometry and transport assays show that phosphorylation of the regulatory
domain relieves this autoinhibition and enhances rMrp2 transport activity. The in vitro data is confirmed in human hepatocyte-like cells, in which inhibition of endogenous kinases also
reduces human MRP2 transport activity. The drug-bound state reveals two probenecid binding sites that suggest a dynamic interplay with autoinhibition. Mapping of the Dubin-Johnson mutations
onto the rodent structure indicates that many may interfere with the transition between conformational states.
MRP2 (multidrug resistance-associated protein, 2) is a primary active transporter of the ATP-binding cassette (ABC) class1. There are nine C-subfamily members in humans with distinct
physiological roles ranging from a chloride channel (the cystic fibrosis transmembrane regulator (CFTR); ABCB7) and a regulator of potassium channels (the sulphonyl urea receptors 1 and 2;
ABCC8 and 9) to exporters of organic anions (MRP1-5).
MRP2 is localised to the apical membranes of polarised cells such as hepatocytes, enterocytes, pneumocytes and proximal tubule cells of the kidney. Loss of MRP2 function manisfests primarily
as a liver disease caused by the failure to export bilirubin glucuronide, the hepatic product of heme breakdown. Failure to eliminate the compound across the canalicular membrane into the
bile ultimately results in jaundice, the primary feature of Dubin–Johnson syndrome, after the conjugated bilirubin is exported back into the circulation by the basolateral MRP32. Over 40
mutations have been identified in the ABCC2 gene, some of which are neutral, others are non-sense or missense mutations causing the absence of the MRP2 protein from the hepatocyte apical
membrane or MRP2 with compromised transport activity, respectively3,4.
Other transport substrates of MRP2 have been identified in vitro and include glutathione- sulfate-, and glucuronide- conjugated metabolites of Phase II metabolism. These include conjugated
leukotriene C4 (conjugated LTC4) and therapeutic anticancer drugs such as paclitaxel and cisplatin5,6,7,8,9,10,11. The physiological relevance of drug transport remains unclear but animal
studies have shown that engineered expression of MRP2 in implanted cancer cells confers resistance to cisplatin12 and in a small clinical study MRP2 expression levels in hepatocellular
cancer patients correlated with a reduction in cisplatin-induced tumour necrosis suggesting that MRP2 is a potential determinant of multidrug resistance13.
The activity of several ABCC transporters are known to be mediated by protein kinases. For example, phosphorylation of MRP1 by the protein kinase CK2 alpha (CK2α) leads to increased efflux
of doxorubicin14, while CFTR is activated by protein kinases C and A to allow ATP-dependent channel opening15,16. In addition to phosphorylation, transport activity can also be modulated by
drug-drug interactions. For MRP2 the uricosuric agent probenecid and the diuretic furosemide have been shown to inhibit the efflux of N-ethylmaleimide glutathione in vitro17. Jaundice is
also a reported off-target side effect of probenecid18.
In this work, we screen for mammalian homologues of human MRP2 and we identify the Rattus norvegicus Mrp2 (rMrp2) as suitable for structural studies (80% sequence identity to its human
ortholog). We determine the cryo-EM structure of rMrp2 in a nucleotide-free state (3.21 Å resolution) and a drug-bound state (3.45 Å resolution) with the uricosuric agent probenecid. The
nucleotide-free state is in an autoinhibited state. Functional data coupled to mass spectrometry-based proteomics identify phosphorylation sites that likely modulate the activation of the
protein and transport data with rMrp2 reconstituted in liposomes show a strong correlation between the phosphorylation state of the transporters and transport activity. This observation is
corroborated in a cellular system using human hepatocytes derived from induced-pluripotent stem cells (iPSCs) which shows a reduction in MRP2 transport activity following inhibition of
protein kinase activity. The probenecid bound structure reveals two drug binding sites within the rMrp2 cavity. The mutations that cause Dubin–Johnson syndrome are mapped onto the rMrp2
structure, which provides insights on how they contribute to MRP2 dysfunction by interfering with conformational changes along the transport cycle rather than substrate binding. Based on
this work, we propose a mechanism for the modulation of the MRP2 activity by phosphorylation and drugs.
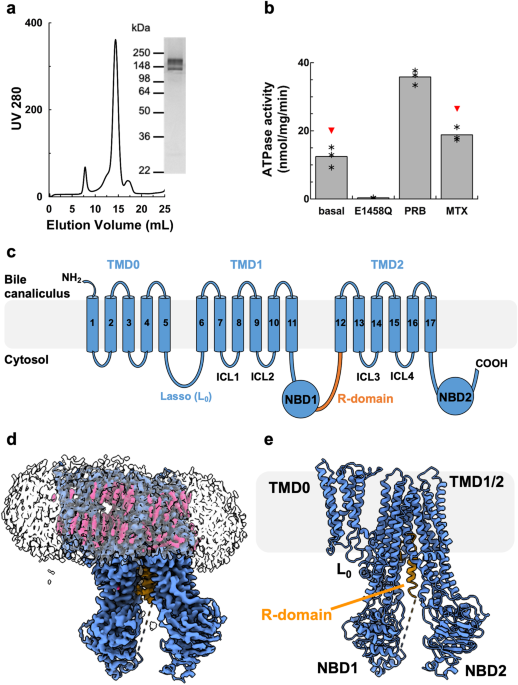
We expressed rMrp2 in Saccharomyces cerevisiae as part of our screen to identify homologues suitable for structural studies19. Extraction of rMrp2 with Lauryl Maltose Neopentyl Glycol (LMNG)
and cholesteryl hemisuccinate (CHS) and further purification into glyco-diosgenin (GDN) resulted in highly pure and monodisperse sample suitable for functional and structural studies (Fig.
1). The recombinant rMrp2 was reconstituted in destabilised liposomes consisting of bovine liver lipid extract, and it displayed a basal ATPase activity of 12.43 ± 3 nmol/mg/min that could
be stimulated by the clinical drugs probenecid and methotrexate by 2.9 and 1.5 fold, respectively17 (Fig. 1). Single-particle cryo-EM analysis of the nucleotide-free rMRP2 resulted in a
three-dimensional reconstruction at an overall resolution of 3.21 Å (Fourier shell correlation (FSC) = 0.143 criterion; Supplementary Fig. 1 and Supplementary Table 1). The maps showed
density with good connectivity and the presence of side chains that could be easily interpreted for model building. The rMrp2 (AF-Q63120-F1) AlphaFold2 model was used as a starting model. We
have built an atomic model for rMrp2 that consists of a small N-terminal transmembrane domain (TMD0) that is linked to two transmembrane domains (TMD), TMD1 and TMD2, by the lasso linker
(L0). TMD0 is composed of a bundle of 5 transmembrane helices (TMs); the interface of TMD0 and TMD1 is occupied by several CHS molecules (not modelled as only part of the sterol moiety was
resolved) (Fig. 1). The L0 linker has been shown to facilitate correct folding and trafficking of ABCC transporters20,21. Each TMD consists of 6 TM helices, enclosing the pocket for
substrates and drugs binding, and they are linked to two nucleotide binding domains (NBDs), NBD1 and NBD2, where ATP binding and hydrolysis provides the free energy required for the
transport cycle completion. NBD1 and NBD2 form an interface with the TMD1 and TMD2 via the intracellular loop 4 (ICL4) and 2 (ICL2), respectively; ICL2 and ICL4 are known as coupling helices
and their role is to transmit conformational changes to the TMDs, associated with either NBD dimerisation or disengagement upon ATP binding or hydrolysis, respectively22. In the absence of
nucleotides, rMrp2 adopts an inward-facing conformation with the TMDs open to the cytosol where substrates and drugs can access it. An unusual feature of the rMrp2 cryo-EM maps was the
presence of helical-like density within the TMD1 and TMD2 interface that corresponds to the regulatory domain (R-domain) (Fig. 2); MRP2 and other members of the ABCC family contain an
R-domain that regulates their transport activity upon phosphorylation by specific kinases (CKII, PKC, PKA and PLK kinases)23. The R-domain is predicted to be partly helical from the
AlphaFold2 model and it links NBD1 to the elbow helix (short helix at the amine-terminal of TM12 and it sits perpendicular to the TMD2), but in the predicted model it was placed at the
interface of
the NBDs and outside the TMD whereas in our structure it sits deep inside the TMD. The R-domain consists of residues Gly863-Thr958. Although, continuous density from NBD1 to the helical
density can be observed, only the helical part, residues Ala895-Lys924, was modelled as the rest of the density is weak and without defined side chains; in addition, there is no density that
links the C-terminus of the R-domain to the elbow helix. The R-domain is buried within the TMDs at the interface of TMs 8, 9, 11 and 17 and it is stabilised by several hydrogen bonds and
Van der Waals interactions (Fig. 2).
a Chromatogram and SDS-PAGE analysis of Size Exclusion Chromatography of purified rMrp2. Representatives from three purifications. b Effect of drugs on the basal ATPase activity of rMrp2
reconstituted in destabilised liposomes. The basal ATPase activity of rMrp2 can be stimulated by 1 mM probenecid (PRB) and 0.1 mM methotrexate (MTX). The E1458Q mutant displays no ATPase
activity. Results are represented as means with individual data points indicated by asterisc, from three independent experiments. Significantly different as estimated by one way ANOVA (p =
2.32 ×10−7) followed by Tukey test p