Insights into the expanding phenotypic spectrum of inherited disorders of biogenic amines
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Inherited disorders of neurotransmitter metabolism are rare neurodevelopmental diseases presenting with movement disorders and global developmental delay. This study presents the
results of the first standardized deep phenotyping approach and describes the clinical and biochemical presentation at disease onset as well as diagnostic approaches of 275 patients from the
registry of the International Working Group on Neurotransmitter related Disorders. The results reveal an increased rate of prematurity, a high risk for being small for gestational age and
for congenital microcephaly in some disorders. Age at diagnosis and the diagnostic delay are influenced by the diagnostic methods applied and by disease-specific symptoms. The timepoint of
investigation was also a significant factor: delay to diagnosis has decreased in recent years, possibly due to novel diagnostic approaches or raised awareness. Although each disorder has a
specific biochemical pattern, we observed confounding exceptions to the rule. The data provide comprehensive insights into the phenotypic spectrum of neurotransmitter disorders. SIMILAR
CONTENT BEING VIEWED BY OTHERS VARIANTS IN _GNAI1_ CAUSE A SYNDROME ASSOCIATED WITH VARIABLE FEATURES INCLUDING DEVELOPMENTAL DELAY, SEIZURES, AND HYPOTONIA Article 20 January 2021 GENETICS
OF GLUTAMATE AND ITS RECEPTORS IN AUTISM SPECTRUM DISORDER Article Open access 16 March 2022 THE PHENOTYPIC SPECTRUM AND GENOTYPE-PHENOTYPE CORRELATIONS IN 106 PATIENTS WITH VARIANTS IN
MAJOR AUTISM GENE _CHD8_ Article Open access 01 October 2022 INTRODUCTION Inherited disorders of neurotransmitter metabolism represent a group of rare neurometabolic diseases. They are
caused by impaired biosynthesis, breakdown or transport of neurotransmitters, or of their essential cofactors, such as tetrahydrobiopterin (BH4). According to the chemical structure of the
primarily affected metabolite they can be classified into distinct groups1 (Table 1): (A) Disorders of biogenic amines (dopamine, serotonin, norepinephrine, epinephrine): (1) Primary
disorders of biogenic amine metabolism: (i) Primary enzyme defects in biogenic amine biosynthesis (aromatic l-amino acid decarboxylase deficiency (AADCD), tyrosine hydroxylase deficiency
(THD)); (ii) Disorders of biogenic amine catabolism (monoamine oxidase A deficiency (MAOAD), dopamine β-hydroxylase deficiency); (iii) Disorders of biogenic amine transport (vesicular
monoamine transporter 2 deficiency, dopamine transporter deficiency (DATD)), (2) Disorders of tetrahydrobiopterin biosynthesis and recycling (autosomal dominant and recessive
GTP-cyclohydrolase deficiency (ad/arGTPCHD), 6-pyruvoyl-tetrahydropterin synthase deficiency (PTPSD), sepiapterin reductase deficiency (SRD), dihydropteridine reductase deficiency (DHPRD),
pterin-4a-carbinolamine dehydratase deficiency), (3) Co-chaperone associated disorders (DNAJC12 deficiency (DNAJC12D)) and (B) Disorders of amino acid neurotransmitters (glycine, glutamate,
serine, γ-aminobutyric acid (GABA)). Manifestations of these disorders mainly involve the central nervous system but other organ systems such as autonomic nervous, hematological or
cardiovascular can also be affected. The clinical phenotype consists of a broad spectrum of symptoms, ranging from mild hypotonia and late-onset movement disorders, to early-onset lethal
encephalopathies. Initial symptoms can appear at any time from the perinatal period to adulthood. Since many clinical symptoms are unspecific or overlap with features seen in other
neurological conditions, such as cerebral palsy, epileptic encephalopathies and hypoxic–ischemic encephalopathy, inherited neurotransmitter disorders are often under-recognized and
misdiagnosed2. Within these disorders only a small group can be detected via newborn screening for phenylketonuria (PKU) while other diseases require selective diagnostic tests leading to
prolonged diagnostic work-up and delayed treatment initiation3. The outcome depends on the underlying disorder, the timing of diagnosis, initiation and type of disease-specific treatment, as
well as long-term compliance to treatment4,5,6,7,8,9,10. Since inherited neurotransmitter disorders are rare disorders, the medical literature is comprised mainly of single case reports,
small case series and retrospective cohort descriptions. The “International Working Group on Neurotransmitter Related Disorders (iNTD)” was founded in 2013 (www.intd-online.org), to overcome
these limitations in clinical and scientific research11. Over the last few years it has steadily grown to include experts from 42 academic and clinical centers from 26 countries. In
December 2014, iNTD set up the first international, longitudinal patient registry. This registry aims to improve our understanding of the natural history, epidemiology, genotype/phenotype
correlations and clinical outcome, and to evaluate diagnostic and therapeutic strategies. In this work, we present the first standardized evaluation of the iNTD patient registry and report
comprehensive insights into pre-, peri- and postnatal presentations of inherited disorders of biogenic amines, as well as effects of initial clinical and biochemical patterns on the
diagnostic process. RESULTS DESCRIPTION OF THE STUDY POPULATION Between January 1st 2015 and May 15th 2020, 429 patients were enrolled in the iNTD patient registry. Of these entries, 350
patients had a diagnosis of biogenic amine disorders. 75 patients who were transferred from the JAKE database on aromatic L-amino acid decarboxylase deficiency
(http://www.biopku.org/home/jake.asp) were not analyzed in this study due to the high number of missing variables of interest. The remaining cohort of patients with disorders of biogenic
amines consisted of 275 patients from 248 families (157 female (57%), from 42 countries: 196 patients born in Europe, 42 in North America, 34 in Asia, three in Central/South America and one
in Africa). 109 patients had primary disorders of biogenic amine metabolism, 161 BH4 deficiencies (BH4Ds) and five patients DNAJC12D (Tables 2 and 3). All diagnoses were confirmed either by
mutational analysis alone or by a combination of specific biochemical tests in CSF, urine and blood (Table 4). There were no patients with dopamine β-hydroxylase deficiency or vesicular
monoamine transporter 2 deficiency. For a reliable explorative analysis, a minimum number of 6 patients was required. MAOAD, DATD and DNAJC12D were included only in the descriptive analysis.
PREGNANCY, DELIVERY AND PERI- AND POSTNATAL OUTCOME Maternal health problems, medications taken during pregnancy and postnatal outcomes are depicted in Tables 2 and 3 and Supplementary
Table 1. None of the patients were prenatally diagnosed. There was no difference in the mode of delivery between the different primary disorders of biogenic amine metabolism (Table 2). Both
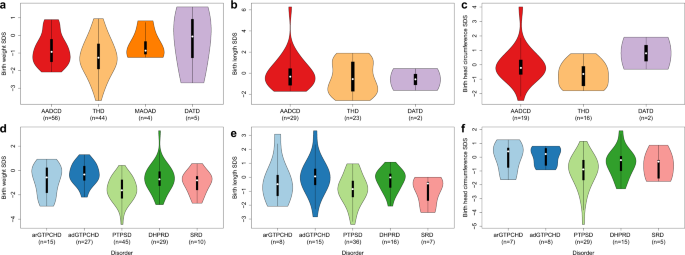
in AADCD and THD a high frequency of small for gestation age (SGA) babies was noted and a remarkably high number of patients with THD had birth length (BL) < 10th percentile (Table 2 and
Fig. 1). In BH4Ds, 27 pregnancies with maternal health issues were reported. Among four mothers affected with adGTPCHD in this group, two mothers did not require any drug treatment despite
having intermittent dystonia. There was no difference with regard to the mode of delivery between arGTPCHD and PTPSD while spontaneous vaginal delivery (SVD) was the most frequent mode of
delivery in adGTPCHD and DHPRD (Table 3). Interestingly, newborns with PTPSD showed the highest rate of symmetrical intrauterine growth restriction (sIUGR) and SGA (Table 3 and Fig. 1).
INITIAL CLINICAL PRESENTATION The most common initial symptoms in AADCD and THD were developmental delay and truncal hypotonia (Fig. 2a). Thermoregulation disorders and oculogyric crises
were prominent in AADCD and occurred more often than in THD (Fig. 3a). Dystonia was similarly frequent in both disease groups. Sleep disturbances were only observed in AADCD. The following
additional symptoms (not listed in Figs. 2a and 3a) were reported in AADCD: gastrointestinal symptoms (_n_ = 7, 12.5%), nasal congestion, (_n_ = 6, 11%), tremor (_n_ = 3, 5%) and hypokinetic
rigid syndrome (_n_ = 3, 5%). In THD, tremor (_n_ = 9, 20.5%), hypokinetic rigid syndrome (_n_ = 3, 7%), gastrointestinal symptoms (_n_ = 3, 7%) and suspected seizures (_n_ = 3, 7%) were
reported. Developmental delay was reported in two out of four patients with MAOAD, epileptic seizures and encephalopathy in one and sleep disorders the other patient. Developmental delay,
hypomimia and drooling were reported in one patient with DATD and dystonia and hypokinesia in another patient. Microcephaly and irritability each were the initial symptoms in other two
patients with DATD. The mean age at onset of first symptoms was 4.3 months across all disorders in this group (range 0–60 months, Table 4). Patients with AADCD were already symptomatic in
the neonatal period and in early infancy while the majority of patients with THD became symptomatic during infancy (between 1 and 15 months, Fig. 3a). Only three out of 33 patients with THD
were symptomatic neonatally with developmental delay and oculogyric crises while two patients showed initial symptoms in childhood (aged 3 and 5 years), one with hypotonia of extremities,
dyskinesia and dystonia, and the other with gait disorder and speech difficulties. The most common initial symptoms among BH4Ds were dystonia, developmental delay and truncal hypotonia (Fig.
2b). Symptom onset occurred most frequently during the neonatal period or infancy in all patients with BH4Ds except for those with adGTPCHD (Table 4 and Fig. 3b). This neonatal/ infantile
initial presentation was typically characterized by developmental delay and (truncal) hypotonia. In contrast, dystonia (61%) and lower extremity hypertonia (13%) were the most frequent
initial symptoms in adGTPCHD, together with gait disorders (_n_ = 6, 16%), orthopedic problems (_n_ = 5, 14%), tremor (_n_ = 3, 8%) and toe walking (_n_ = 3, 8%, not listed in Figs. 2b and
3b). Dystonia was more frequently seen in arGTPCHD (22%) than in PTPSD (11%), DHPRD (5%) and SRD (7%) in an age-independent analysis (Fig. 3b, age-dependent analysis). Seizures were reported
only in PTPSD (5%) and DHPRD (11%) among all BH4Ds. As additional initial symptoms, hypokinetic rigid syndrome was reported in PTPSD (_n_ = 3, 5%) and DHPRD (_n_ = 3, 8%) and failure to
thrive in PTPSD (_n_ = 3, 5%). Of note is the higher frequency of oculogyric crises (21%), and sleep disorders (14%) in SRD compared to other BH4Ds (0–5% and 0–6% respectively;
age-independent analysis). Developmental delay alone was reported in one patient with DNAJC12D, together with oculogyric crises in another patient and behavioral problems in two patients.
DIAGNOSTIC WORK-UP AND DIAGNOSTIC DELAY The latency to diagnosis was comparably long for AADCD (mean = 41 months, range 0–386 months) and THD (mean = 45 months, range −6–361 months) (Fig.
4a). Patients with AADCD born before 2009 had a statistically significant longer latency to diagnosis (mean 68.1 months) than those born after 2009 (mean 7.3 months, _p_ = 0.00041, WMW-test)
(Fig. 4b). In the group of patients with THD, this pattern was also found for patients born before and after 2005, respectively (mean diagnostic delay of 89.8 months vs. 14.4 months, _p_ =
0.00041, WMW-test). Investigating the effect of symptoms on age at diagnosis or on the diagnostic delay, we found that presentation with hypotonia, seizures, encephalopathy, microcephaly,
sleep disturbances or thermoregulation disorders was associated with earlier age at diagnosis (3.2 years) and with less diagnostic delay (30 months), than presentation with dystonia,
dyskinesia, hypoglycemia or developmental delay (diagnosis age 4.9 years, _t_ (88.14) = −1.68; _p_ = 0.1; diagnostic delay 45 months, _t_ (175.43) = −1.89; _p_ = 0.06; Fig. 5a and b). Since
disorders presenting with hyperphenylalaninemia (HPA), arGTPCHD, PTPSD and DHPRD, can be detected by newborn screening (NBS) for phenylketonuria, they would be expected to be diagnosed at a
younger age and with less delay than those without HPA. In our study cohort, diagnostic work-up as well as diagnostic delay varied strongly depending on the occurrence of HPA (Table 4 and
Fig. 4a). In patients with arGTPCHD, PTPSD and DHPRD, HPA on NBS led to significantly shorter diagnostic delay than in cases without HPA or without any NBS performed (mean 3.1 vs. 39.6
months, _t_ (24.05) = 2.99; _p_ = 0.006, Fig. 4c). There was no significant difference in diagnostic delay between the latter group vs. those BH4Ds without HPA and AADCD and THD (mean 39.6
vs. 76.7 vs. 51 months, _t_ (2) = 1.89; _p_ = 0.15). Disorders without HPA (adGTPCHD and SRD) were mainly diagnosed by selective screening after onset of symptoms or by high-risk family
screening (HRFs). In our study, the majority of patients with adGTPCHD were symptomatic in childhood and had a mean diagnostic delay of 60 months (range 1–245 months). Most patients with SRD
were symptomatic in the first 6 months of life, however, this group in particular showed a prominently prolonged latency to diagnosis with an average duration of 112 months (range 12–316
months, Table 4, Fig. 4a). SRD patients born after 2009 had a significantly shorter diagnostic delay than those born before (mean 140 months, vs. 29.9 months, _p_ = 0.021, WMW-test). For
adGTPCHD no significantly discriminating date was found (Fig. 4b). Seven patients with arGTPCHD had normal NBS results without HPA. Eight patients presented with HPA on NBS but in six of
these eight cases the diagnostic work-up was initiated later (Table 4). In patients with arGTPCHD who were diagnosed via selective screening, the mean diagnostic delay in the group with HPA
on NBS (7.8 months) differed from the other group with normal results on NBS (21.5 months) without reaching statistical significance (_t_ (7.39) = −1.78; _p_ = 0.11, Fig. 4d). Only in the
PTPSD group did all the available NBS results demonstrate HPA (Table 4). While the vast majority of the patients with DHPRD (30/37) had HPA on NBS, one patient surprisingly had a normal NBS
and presented normal phenylalanine levels in plasma but high levels in CSF on repeated measurements. This patient was homozygous for a new variant in the _QDPR_ gene (NM_000320.3, c.466 G
> A, p. Ala156Thr). There were 17 DHPRD patients whose diagnosis was established late despite having HPA on NBS and who had a longer mean diagnostic delay (6 months) than those who were
diagnosed by specific work-up immediately following the detection of HPA on NBS (-4.5 months, _t_ (16.44) = −1.87; _p_ = 0.08, Fig. 4e). Negative values for the diagnostic delay in PTPSD and
DHPRD in Fig. 4a are explained by an early diagnosis via NBS or HRFs before onset of symptoms. The lowest values for the mean diagnostic delay (8 months) and for the maximum diagnostic
delay (86 months) were recorded in DHPRD. We identified the birth year 1993 as the most strongly discriminating and statistically significant time point regarding changes in the latency to
diagnosis for PTPSD (mean 86.6 months, SD 88.6 months vs. mean 3.4 months, SD 17.3 months, p = 0.028, WMW-test, Fig. 4b). In the remaining BH4Ds, we could detect a trend around the years
1999 for arGTPCHD and 2012 for DHPRD but these dates did not reach statistical significance. Truncal hypotonia, upper limb hyper-/or hypotonia, developmental delay, epilepsy, encephalopathy,
microcephaly, thermoregulation disorders, oculogyric crises, dyskinesia or hypoglycemia were associated with earlier age at diagnosis (2.7 years) than lower limb hypo-/or hypertonia (4.7
years) or dystonia and sleep disorders (8.8 years; ANOVA; _F_ (2,165) = 14.89; _p_ = 0.16 for 2.7 vs 4.7 years; _p_ = 0.0000005 for 2.7 vs 8.8 years; _p_ = 0.02 for 4.7 vs. 8.8 years). Other
than in primary disorders of biogenic amine metabolism, developmental delay, tone abnormalities in upper limb and trunk as well as epilepsy were associated with a shorter diagnostic delay
(28 months) than oculogyric crises, dystonia, lower limb tone abnormalities and sleep problems (47 months, _t_ (117.73) = −2.15; _p_ = 0.03, Fig. 5c, d) in BH4Ds. INITIAL BIOCHEMICAL
PRESENTATION Diagnostically relevant and disease-specific constellations of biochemical parameters are presented in Fig. 6. Along with typical changes in biogenic amines (i.e. reduced
homovanillic acid (HVA) and 5-hydroxyindolacetic acid (5-HIAA), elevated 3-O-methyl-Dopa (3-OMD), levodopa (L-Dopa) and 5-hydroxytryptophan (5-HTP)), abnormalities of tetrahydrobiopterin and
neopterin in CSF were observed in some AADCD patients (Fig. 6) Urinary vanillactic acid was not reported as an initial diagnostic parameter in our AADCD cohort. In THD, HVA and the ratio
HVA/5-HIAA were decreased in almost all samples, while 5-HTP and 5-HIAA were typically normal. Prolactin in plasma showed high variability in both diseases (Fig. 6). Phenylalanine (Phe) was
normal or high in CSF and plasma in arGTPCHD while it was predominantly normal in both blood and CSF in adGTPCHD (Fig. 6). Phe was high in both CSF and in plasma in PTPSD and DHPRD. In SRD,
Phe was increased in CSF while being normal in plasma, in line with previous reports12. Pterin disturbances were reported in CSF and in urine for adGTPCHD. Results on pterins in dried blood
spots (DBS) in adGTPCHD were not available. 7-8-dihydrobiopterin in CSF was determined rarely but three measurements in DHPRD and two in SRD were high while being always normal in other BH4
disorders. HVA and 5-HIAA were more frequently decreased in PTPSD, DHPRD and SRD than in ad/arGTPCHD. In PTPSD and DHPRD prolactin was elevated in 44% and 82% of cases, respectively, while
it was normal in almost all other BH4Ds. Decreased 5-methyltetrahydrofolate in CSF was reported only in DHPRD, except in one case with arGTPCHD and three cases with PTPSD. FATAL OUTCOMES
Three patients with AADCD and one with THD died during the study period. Death occurred at 2.4, 2.6 and 19.8 years of life in the AADCD patients. One patient died of pneumonia while in the
remaining two cases the cause was unknown. The THD patient died at 13 years of age because of an acute lower respiratory tract infection. DISCUSSION The evaluation of 275 patients with
disorders of biogenic amines (224 new and 51 previously published cases) that were analyzed using a standardized longitudinal approach revealed new phenotypic aspects of the initial clinical
and biochemical presentation, peri- and postnatal courses as well as diagnostic work-up. We present an increased incidence of prematurity in AADCD and of SGA in THD and in PTPSD. Patients
with PTPSD were also prone to sIUGR and congenital microcephaly. We report one patient with DHPRD without HPA on NBS. We confirm the significant impact of HPA detection on NBS on the
diagnostic work-up in a group of BH4Ds. Furthermore, we present the association of specific symptoms, such as oculogyric crises, dystonia, sleep and thermoregulation disorders, with age at
diagnosis and diagnostic delay. Pregnancies in both main disease groups were rarely complicated by medical problems. The issues described were most likely due to the pregnancy itself and not
to the fetal disease. Exceptions were those cases in which mothers were affected by adGTPCHD, consistent with previous literature13. First symptoms of disorders of biogenic amines typically
occur in the neonatal period or in infancy. Our data on the anthropometrical values at birth raise questions about prenatal disease manifestation. AADCD (18%) and SRD (21%) showed an
increased rate of prematurity in our study compared to the global incidence of prematurity that is estimated as 9.6% ranging from 6.2% in Europe to 9.1 % in Asia, 10.6 % in North America and
11.9% in Africa14. Various causal factors such as fetal or maternal health conditions along with genetic, environmental, behavioral and socioeconomic factors as well as the differences in
availability of preventive interventions between developed and developing countries influence the estimated rates. Since most of the patients in this study were born in Europe, North America
or Asia, the background preterm birth rate may be expected to be between 6.2% and 10.6%. While the rate in SRD should be critically interpreted due the small number of cases, our data
document an increased rate of prematurity in AADCD. Additionally, the risk of SGA at birth is higher in PTPSD and THD (56% and 49%, respectively) compared to the considerable variation in
the prevalence of infants born SGA, ranging from 4.6–15.3 % across Europe and 5.3% in east Asia to 41.5% in south Asia15,16. AADCD, arGTPCHD, DHPRD and SRD also show elevated but not as high
SGA rates. While neonates with PTPSD are also prone to sIUGR and congenital microcephaly (24%), we could not observe any trend towards microcephaly in DHPRD in contrast to the reported 25%
rate in a historical cohort17. The observation that PTPSD patients were at high risk for prematurity could not be confirmed in our cohort but the detection of SGA and sIUGR are in line with
the previously reported tendency to have very low birth weight (BW < 1500 gram5,18). To date, there are no previous reports of relevant changes in birth metrics in newborns with THD. In
previous publications the clinical phenotype of THD was divided into type A and severe type B19, with higher frequency of perinatal abnormalities, including prematurity in patients with type
B. In our iNTD study group of THD, we could not find a clear difference that would justify a differentiation into two types of severity. Furthermore, THD patients did not show an increased
frequency of prematurity compared to the overall incidence as mentioned above. Perinatal abnormalities, postnatal problems, achievement of gross motor milestones including walking without
assistance and medication varied regardless of the age of initial symptoms. Therefore, we propose to abandon this classification. These observed peri- and postnatal changes indicate that
impairments of biogenic amine neurotransmitter metabolism and their effects on the fetus start during pregnancy. Embryonic lethality was reported in _TH_, _DßH_ and homozygous
_GCH1_knock-out mice previously20,21,22. In mice, a rescue until term is possible with L-Dopa, BH4 or dihydroxyphenylserine supplementation indicating that noradrenaline, dopamine and BH4
are essential for fetal development. Homozygous _PTPS_ and _Spr_knock-out mice, and _GCH1_knock-in mice are born visibly normal, have growth retardation postnatally and die after 48 h to a
few weeks of age23,24,25,26. The _Qdpr__−/−_ mice are indistinguishable from their wildtype littermates and show normal growth27. Furthermore, genetically rescued _PTPS_ knock-out,
_TH_knock-in and _AADC_knock-in murine models are born without obvious morphological abnormality and survive but show growth retardation23,28,29. These animal models demonstrate that BH4
regulates catecholamine synthesis through altering TH protein levels and that the postnatally expected increase of dopamine and TH protein concentration in the brain is disrupted by BH4
deficiency23,25. Maternal compensation of BH4 and dopamine deficiency as well as postnatal rescue has been demonstrated to be possible but limited20,21,22,24,25,30. Further studies will be
needed to elucidate the disease-related pre- and postnatal findings of our study and the effectiveness and limitations of maternal metabolic compensation. Nonspecific symptoms, such as
feeding problems and hyperbilirubinemia were the most common postnatal problems in our study. A small group of patients with arGTPCHD presented with tremor, jitteriness, irritability and
some patients with AADCD showed temperature instability and/or hypoglycemia postnatally that could be interpreted as disease-related. Following the postnatal period, the clinical
presentation varied between diseases, although the symptoms are caused by a similar pathophysiological mechanism with the disruption in dopaminergic and/or serotoninergic neurotransmission.
We showed that while AADCD presented with a variety of non-motor and motor symptoms31,32, THD had an initial clinical picture clearly dominated by motor symptoms in addition to developmental
delay starting on average in early infancy. In the case of BH4Ds our results are similar to previous reports5,17,33,34. Seizures were typical in DHPRD and PTPSD among BH4Ds while sleep
disorders were especially frequent in SRD. These observations cannot be explained solely by any evidence but some hypotheses can be generated based on previous reports on different pathways:
(1) Decreased BH4 concentrations and elevated levels of 7,8-dihydrobiopterin (BH2) in DHPRD and SRD, in the latter together with elevated sepiapterin, lead to disturbances in intracellular
BH4:BH2 ratio that codetermines uncoupling of endothelial NOS, resulting in generation of oxygen radicals35,36. Although the impact of these perturbations on the clinical picture remains
unclear, interactions between NO concentration and NOS activity and epileptic discharges37,38 as well as sleep initiation and maintenance39 have been postulated. (2) The secondary cerebral
folate deficiency occurring frequently in DHPRD40 may result in severe epileptic encephalopathy41,42. (3) A disturbed melatonin homeostasis, documented by low urinary sulphatoxymelatonin
levels in patients with SRD could provide another pathophysiological link43. Our data demonstrate that if recognized correctly, some disease-related symptoms could raise clinical suspicion
and facilitate prompt diagnosis. Sleep and thermoregulation disturbances were associated with an earlier age at diagnosis and shorter latency to diagnosis, especially in AADCD. Oculogyric
crises were associated with a longer diagnostic delay, although they correlated with an earlier age at diagnosis. This observation implies that oculogyric crises occur early and that correct
recognition of the symptom could potentially shorten the diagnostic delay. Developmental delay was associated with a later age at diagnosis and a longer diagnostic delay in primary
disorders of biogenic amine metabolism in contrast to BH4Ds. This could be explained on one hand by the fact that developmental delay is a rather nonspecific symptom and on the other hand
that it could be recognized more easily and earlier by a physician or parents. We could furthermore show an association of dystonia with higher age at diagnosis and a longer latency to
diagnosis, probably due to the broad spectrum of differential diagnoses of dystonia44. Initial biochemical profiles largely revealed disease-specific patterns but our data showed occasional
atypical biochemical findings that underline the importance of a careful interpretation of pterins and biogenic amines together with the clinical picture. For example, some patients with
AADCD had abnormal concentrations of tetrahydrobiopterin and neopterin in CSF, in contrast to previous literature that emphasized the importance of normal pterin findings in AADCD32.
Therefore, one should be aware of potential secondary disturbances most likely without clinical significance. Our data confirm that arGTPCHD can present with or without HPA but we also found
some exceptions to the rule about which disorders present with HPA. Our registry contains one patient with a new variant in the _QDPR_ gene showing repeatedly normal phenylalanine levels in
plasma but high levels in CSF. This observation needs to be studied with further functional analyses. Furthermore, we observed that ad/arGTPCHD and PTPSD may also have increased or normal
HVA and 5-HIAA in CSF in contrast to DHPRD and SRD, in which HVA and 5-HIAA were mainly decreased. Pterin profiles tend to be consistent and disease-specific in BH4Ds, except in DHPRD. Also,
in line with previous reports, we observe that pterin measurements in urine and CSF are more sensitive than in DBS or plasma3,17. Hyperprolactinemia is frequently used as a marker of
central dopaminergic deficiency, but normal prolactin levels do not exclude a neurotransmitter disorder45. Our data support this statement, as prolactin levels were scattered from low to
high in disorders where cerebral dopaminergic deficiency with decreased HVA was documented. When interpreting blood prolactin, other causes for hyperprolactinemia must also be considered46.
Our results also provide additional insights into the inter-dependence of disease-specific diagnostic delay and diagnostic tools. The suspected diagnoses were confirmed most frequently by
mutational analysis alone or in combination with specific biochemical tests. Those diseases that had abnormal NBS due to HPA had a significant shorter diagnostic delay. Overall, DHPRD could
be diagnosed fastest, probably due to easily accessible DHPR activity measurement in DBS. In arGTPCHD and DHPRD a tendency to diagnose still asymptomatic children before disease onset,
following a pathological NBS result was found. This underlines the importance of a complete, systematic and timely diagnostic work-up of every HPA as recommended in the recently published
guidelines17. It is important to note, since arGTPCHD and DHPRD rarely may show normal phenylalanine concentrations on NBS, a diagnostic re-evaluation should be performed if there is strong
clinical suspicion. As adGTPCHD and SRD are not detectable on NBS for PKU, the finding of a prolonged diagnostic delay in these disorders is not unexpected. In our study, SRD had the highest
latency to diagnosis as reported previously5,33. This is presumably caused by the absence of an easily accessible biochemical marker and the challenging clinical picture of SRD. In
addition, characteristics of SRD such as oculogyric crises and sleep disorders may remain undetected or be misinterpreted. Complementary to the literature, our study demonstrated that
patients with adGTPCHD may show symptoms as early as 6 months of age, and that the mean latency to diagnosis is approximately 5 years, shorter than the previously reported latency of 13–16
years34,47. Considering that PTPSD was first described in the 1980s48, AADCD and THD in the 1990s49,50 and SRD in 200151, our observation of a tendency to a shortened diagnostic delay in
correlation with more recent birth dates confirms that enhanced knowledge and awareness of these diseases, efforts to establish a standardized algorithm for diagnostic work-up, and broadened
availability of (genetic) diagnostic tools all likely reduce patients’ diagnostic odyssey. The latter aspect can be supported by the fact that the implementation of next-generation
sequencing-based diagnostics using approaches such as targeted resequencing, whole-exome and whole-genome sequencing has strongly changed clinical genetics52,53. Since the first whole-exome
sequencing proof of concept experiments in 200954,55, the discovery of disease-causing genes using these technologies has increased rapidly. The impact of modern genetic diagnostic tools on
the length of the diagnostic process in patients suspected of having rare genetic conditions has been52,56 and will be an interesting subject for further studies. In our study no patient
with BH4D was reported as deceased, in contrast to the mortality rate in PTPSD and DHPRD in a previous retrospective study5. This can be explained by the size of our study population and by
the fact that only 10 out of 161 patients with BH4Ds were born before the establishment of newborn screening programs in the 1980s. Based on the fact that the inclusion criteria and
requirements for patient enrollment into the iNTD registry were strictly defined and bound to complex ethical standards, the number of patients enrolled is not comparable with earlier
retrospective registry studies (e.g. JAKE; http://www.biopku.org/home/jake.asp, BIODEF57; http:// www.biopku.org). Our registry provides data depending on the participating centers and does
not present the overall incidence of the diseases worldwide. In addition, the medical history module of the registry analyzed in our study is the only part, which gathers data summarized by
treating physicians, parents, caregivers as well as patients. Therefore, some information was retrospective or missing. Following our observation of a high incidence of prematurity and SGA
in some diseases, a revision of the exclusion criterion “BW < 1500 gram” will be proposed to the iNTD steering committee to avoid an underestimation of patients who are born preterm or
with very low birth weight. The results of our study reinforce that international cooperation and patient registries are essential for a better understanding of rare diseases as well as for
harmonization of diagnostic algorithms and standards of patient care in inherited disorders of neurotransmitter metabolism. In conclusion, we describe comprehensive insights into pre-, peri-
and postnatal presentations of inherited disorders of biogenic amines, as well as specific clinical and biochemical patterns affecting the diagnostic process. Our results emphasize the
importance of recognizing the potential early signs and of careful and systematic clinical evaluation to improve diagnostic approaches in these rare neurodevelopmental diseases. These
observations should serve as a basis for further studies on the evolving phenotypic spectrum in disorders of biogenic amines. METHODS THE INTERNATIONAL WORKING GROUP ON NEUROTRANSMITTER
RELATED DISORDERS (INTD) The iNTD patient registry, which is web-based and password-protected (https://www.intd-registry.org), was approved by the Institutional Research Ethics Board (IRB)
Heidelberg University Hospital (S-471/2014, registered German Clinical Trials Register, https://www.drks.de, DRKS00007878) on December 22nd 2014 and subsequently by all contributing
centers11. All procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2013. Written informed consent was obtained from all study participants or their
legal guardians. iNTD was founded without any industry involvement or sponsorship. Exclusion criteria of the iNTD patient registry: Patients with severe comorbidities, e.g. Down syndrome,
intraventricular hemorrhage (°III-IV) in the newborn period, very low birth weight (<1500 gram), kernicterus, embryofetal disease due to maternal alcohol or drug abuse. DEFINITIONS
Gestational age was calculated based on completed weeks of gestation. Preterm pregnancy was defined according to the International Classification of Disease as delivery before 37 completed
(<37 + 0) weeks of gestation58,59. Small for gestational age (SGA) was defined as birth weight (BW) below the 10th percentile. Microcephaly was defined as head circumference at birth
(BHC) below the 3rd percentile60. Symmetrical intrauterine growth restriction (sIUGR) was used as only referring to parameters available postnatally and was defined as BW, BHC and birth
length (BL) below 10th percentile61. We defined the neonatal period as the first 30 days of life. Infancy referred to 31 days to 24 months, childhood to 3–12 years, adolescence 13–18 years
and adulthood older than 18 years. Initial symptoms are the first clinical findings that are considered to be disease related, observed by the physician, parents or the patient and can be
both objective and subjective. The evaluation of the initial symptoms was based on retrospective data. The following symptoms were available in the medical history form in a controlled
vocabulary: encephalopathy, developmental delay (psychomotor retardation), microcephaly, seizures, muscular symptoms, dystonia, dyskinesia, oculogyric crisis, thermoregulation disorders,
sleep disorders and hypoglycemic episodes. Localization and type of muscular symptoms as well as type, frequency and length of seizures could be specified. Free text boxes were available for
additional symptoms. The frequency of initial symptoms was analyzed in both age-dependent and age-independent manner. Additional symptoms included only in free text boxes were described
age-independent. Selective screening stands for a diagnostic process following onset of clinical symptoms. High-risk family screening describes a targeted diagnostic work-up initiated due to
a confirmed disease case in the family, before or after onset of an individual’s clinical symptoms. To describe the initial biochemical presentation (at time of diagnosis), we analyzed the
most frequently measured parameters among 30 biochemical parameters listed in the registry. STATISTICAL ANALYSIS Statistical analyses were performed in R (version 4.0.2). Numeric variables
were compared between two independent groups with Wilcox-Mann-Whitney (WMW) test, or _t_-test with Welch correction, setting _P_ < 0.05 as significant. Analysis of variance (ANOVA) was
used to compare numeric variables between more than two groups. The Benjamini-Hochberg adjustment was applied to correct type 1 error, when applicable. No a-priori hypotheses were tested and
therefore all p-values had descriptive character. Classification and regression trees (CART) were used to identify constellations of symptoms that might have an impact on age at diagnosis
or diagnostic delay (= age at diagnosis - age at initial symptom) for different diseases. Standard deviation scores (SDSs) for anthropometric variables at birth were computed according
Fenton et al.62 REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY All data supporting
the findings described in this manuscript are not publicly available due to existing data protection laws but are available from the corresponding author (T.O.) upon reasonable request and
within the limitations of the informed consent. All requests for raw and analyzed data will be reviewed by the iNTD executive board and iNTD members within 72 h. Data ownership is maintained
by the members of the iNTD network. All participating iNTD members approved this study. Source data for all the figures, tables 2–4andsupplementary table 1 are provided with the paper. Data
transferred from the JAKE database (http://www.biopku.org/home/jake.asp) was not analyzed in this study. Source data are provided with this paper. REFERENCES * Nestler, E. J., Hyman, S. E.,
Holtzman, D. M. & Malenka, R. C. Molecular neuropharmacology: a foundation for clinical neuroscience (2015). * Ng, J., Papandreou, A., Heales, S. J. & Kurian, M. A. Monoamine
neurotransmitter disorders–clinical advances and future perspectives. _Nat. Rev. Neurol._ 11, 567–584 (2015). Article CAS PubMed Google Scholar * Jung-Klawitter, S. & Kuseyri
Hubschmann, O. Analysis of catecholamines and pterins in inborn errors of monoamine neurotransmitter metabolism-from past to future. _Cells_ 8, 867 (2019). * Jaggi, L. et al. Outcome and
long-term follow-up of 36 patients with tetrahydrobiopterin deficiency. _Mol. Genet. Metab._ 93, 295–305 (2008). Article CAS PubMed Google Scholar * Opladen, T., Hoffmann, G. F. &
Blau, N. An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia. _J. Inherit. Metab. Dis._ 35, 963–973 (2012). Article CAS PubMed
Google Scholar * Ponzone, A., Ferraris, S., Baglieri, S. & Spada, M. Treatment of tetrahydrobiopterin deficiencies. In _PKU and BH4: Advances in Phenylketonuria and
Tetrahydrobiopterin_ (ed. Blau, N.) 612–637 (SPS Verlagsgesellschaft, Heilbronn, 2006). * Brun, L. et al. Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency.
_Neurology_ 75, 64–71 (2010). Article CAS PubMed Google Scholar * Serrano, M., Perez-Duenas, B., Montoya, J., Ormazabal, A. & Artuch, R. Genetic causes of cerebral folate
deficiency: clinical, biochemical and therapeutic aspects. _Drug Discov. Today_ 17, 1299–1306 (2012). Article CAS PubMed Google Scholar * Pearl, P. L. et al. Clinical spectrum of
succinic semialdehyde dehydrogenase deficiency. _Neurology_ 60, 1413–1417 (2003). Article CAS PubMed Google Scholar * Swanson, M. A. et al. Biochemical and molecular predictors for
prognosis in nonketotic hyperglycinemia. _Ann. Neurol._ 78, 606–618 (2015). Article CAS PubMed PubMed Central Google Scholar * Opladen, T. et al. The International Working Group on
Neurotransmitter related Disorders (iNTD): a worldwide research project focused on primary and secondary neurotransmitter disorders. _Mol. Genet Metab. Rep._ 9, 61–66 (2016). Article PubMed
PubMed Central Google Scholar * Zorzi, G. et al. Detection of sepiapterin in CSF of patients with sepiapterin reductase deficiency. _Mol. Genet. Metab._ 75, 174–177 (2002). Article CAS
PubMed Google Scholar * Kuseyri, O. et al. Pregnancy management and outcome in patients with four different tetrahydrobiopterin disorders. _J. Inherit. Metab. Dis._ 41, 849–863 (2018).
Article CAS PubMed Google Scholar * Beck, S. et al. The worldwide incidence of preterm birth: a systematic review of maternal mortality and morbidity. _Bull. World Health Organ._ 88,
31–38 (2010). Article PubMed Google Scholar * Lee, A. C. et al. National and regional estimates of term and preterm babies born small for gestational age in 138 low-income and
middle-income countries in 2010. _The Lancet_. _Glob. Health_ 1, e26–e36 (2013). Google Scholar * Ruiz, M. et al. Mother’s education and the risk of preterm and small for gestational age
birth: a DRIVERS meta-analysis of 12 European cohorts. _J. Epidemiol. Community Health_ 69, 826–833 (2015). Article PubMed Google Scholar * Opladen, T. et al. Consensus guideline for the
diagnosis and treatment of tetrahydrobiopterin (BH4) deficiencies. _Orphanet J. Rare Dis._ 15, 126 (2020). Article PubMed PubMed Central Google Scholar * Smith, I. & Dhondt, J.-L.
Birthweight in pateints with defective biopterin metabolism. _Lancet_ 325, 818 (1985). Article Google Scholar * Willemsen, M. A. et al. Tyrosine hydroxylase deficiency: a treatable
disorder of brain catecholamine biosynthesis. _Brain_ 133, 1810–1822 (2010). Article PubMed Google Scholar * Douglas, G. et al. A requirement for Gch1 and tetrahydrobiopterin in embryonic
development. _Dev. Biol._ 399, 129–138 (2015). Article CAS PubMed PubMed Central Google Scholar * Thomas, S. A., Matsumoto, A. M. & Palmiter, R. D. Noradrenaline is essential for
mouse fetal development. _Nature_ 374, 643–646 (1995). Article ADS CAS PubMed Google Scholar * Zhou, Q. Y., Quaife, C. J. & Palmiter, R. D. Targeted disruption of the tyrosine
hydroxylase gene reveals that catecholamines are required for mouse fetal development. _Nature_ 374, 640–643 (1995). Article ADS CAS PubMed Google Scholar * Homma, D. et al. Partial
biopterin deficiency disturbs postnatal development of the dopaminergic system in the brain. _J. Biol. Chem._ 286, 1445–1452 (2011). Article CAS PubMed Google Scholar * Jiang, X. et al.
A novel GTPCH deficiency mouse model exhibiting tetrahydrobiopterin-related metabolic disturbance and infancy-onset motor impairments. _Metabolism_ 94, 96–104 (2019). Article CAS PubMed
Google Scholar * Sumi-Ichinose, C. et al. Catecholamines and serotonin are differently regulated by tetrahydrobiopterin. A study from 6-pyruvoyltetrahydropterin synthase knockout mice. _J.
Biol. Chem._ 276, 41150–41160 (2001). Article CAS PubMed Google Scholar * Yang, S. et al. A murine model for human sepiapterin-reductase deficiency. _Am. J. Hum. Genet_ 78, 575–587
(2006). Article CAS PubMed PubMed Central Google Scholar * Xu, F. et al. Disturbed biopterin and folate metabolism in the Qdpr-deficient mouse. _FEBS Lett._ 588, 3924–3931 (2014).
Article CAS PubMed Google Scholar * Korner, G. et al. Brain catecholamine depletion and motor impairment in a Th knock-in mouse with type B tyrosine hydroxylase deficiency. _Brain_ 138,
2948–2963 (2015). Article PubMed Google Scholar * Lee, N. C. et al. Regulation of the dopaminergic system in a murine model of aromatic L-amino acid decarboxylase deficiency. _Neurobiol.
Dis._ 52, 177–190 (2013). Article CAS PubMed Google Scholar * Elzaouk, L. et al. Dwarfism and low insulin-like growth factor-1 due to dopamine depletion in Pts-/- mice rescued by feeding
neurotransmitter precursors and H4-biopterin. _J. Biol. Chem._ 278, 28303–28311 (2003). Article CAS PubMed Google Scholar * Pearson, T. S., et al. AADC deficiency from infancy to
adulthood: symptoms and developmental outcome in an international cohort of 63 patients. _J. Inherit. Metab. Dis._ 43, 1121 (2020). * Wassenberg, T. et al. Consensus guideline for the
diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. _Orphanet J. Rare Dis._ 12, 12 (2017). Article PubMed PubMed Central Google Scholar * Friedman, J. et
al. Sepiapterin reductase deficiency: a treatable mimic of cerebral palsy. _Ann. Neurol._ 71, 520–530 (2012). Article CAS PubMed Google Scholar * Lopez-Laso, E. et al. Dopa-responsive
infantile hypokinetic rigid syndrome due to dominant guanosine triphosphate cyclohydrolase 1 deficiency. _J. Neurol. Sci._ 256, 90–93 (2007). Article CAS PubMed Google Scholar *
Crabtree, M. J. & Channon, K. M. Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. _Nitric oxide: Biol. Chem._ 25, 81–88 (2011). Article CAS
Google Scholar * Gao, L. et al. Sepiapterin reductase regulation of endothelial tetrahydrobiopterin and nitric oxide bioavailability. _Am. J. Physiol. Heart Circ. Physiol_. 297, H331-H339
(2009). * Jayakumar, A. R., Sujatha, R., Paul, V., Puviarasan, K. & Jayakumar, R. Involvement of nitric oxide and nitric oxide synthase activity in anticonvulsive action. _Brain Res.
Bull._ 48, 387–394 (1999). Article CAS PubMed Google Scholar * Kovacs, R. et al. Endogenous nitric oxide is a key promoting factor for initiation of seizure-like events in hippocampal
and entorhinal cortex slices. _J. Neurosci._ 29, 8565–8577 (2009). Article CAS PubMed PubMed Central Google Scholar * Greene, R. W. Role for neuronal nitric oxide synthase in sleep
homeostasis and arousal. _PNAS_ 110, 19982–19983 (2013). Article ADS CAS PubMed PubMed Central Google Scholar * Xu, F. et al. Disturbed biopterin and folate metabolism in the
Qdpr-deficient mouse. _FEBS Lett._ 588, 3924–3931 (2014). Article CAS PubMed Google Scholar * Pope, S., Artuch, R., Heales, S. & Rahman, S. Cerebral folate deficiency: analytical
tests and differential diagnosis. _J. Inherit. Metab. Dis._ 42, 655–672 (2019). Article CAS PubMed Google Scholar * Kuseyri Hübschmann, O., et al. Brain MR patterns in inherited
disorders of monoamine neurotransmitters: an analysis of 70 patients. _J. Inherit. Metab. Dis._ 44, 1070 (2021). * Batllori, M. et al. Urinary sulphatoxymelatonin as a biomarker of serotonin
status in biogenic amine-deficient patients. _Sci. Rep._ 7, 14675–14675 (2017). Article ADS PubMed PubMed Central CAS Google Scholar * Assmann, B., Surtees, R. & Hoffmann, G. F.
Approach to the diagnosis of neurotransmitter diseases exemplified by the differential diagnosis of childhood-onset dystonia. _Ann. Neurol_. 54, S18–S24 (2003). * Aitkenhead, H. &
Heales, S. J. Establishment of paediatric age-related reference intervals for serum prolactin to aid in the diagnosis of neurometabolic conditions affecting dopamine metabolism. _Ann. Clin.
Biochem_ 50, 156–158 (2013). Article CAS PubMed Google Scholar * Capozzi, A., Scambia, G., Pontecorvi, A. & Lello, S. Hyperprolactinemia: pathophysiology and therapeutic approach.
_Gynecol. Endocrinol._ 31, 506–510 (2015). Article CAS PubMed Google Scholar * Tadic, V. et al. Dopa-responsive dystonia revisited: diagnostic delay, residual signs, and nonmotor signs.
_Arch. Neurol._ 69, 1558–1562 (2012). Article PubMed Google Scholar * Niederwieser, A. et al. “Peripheral” tetrahydrobiopterin deficiency with hyperphenylalaninaemia due to incomplete
6-pyruvoyl tetrahydropterin synthase deficiency or heterozygosity. _Eur. J. Pediatr._ 146, 228–232 (1987). Article CAS PubMed Google Scholar * Hyland, K. & Clayton, P. T. Aromatic
amino acid decarboxylase deficiency in twins. _J. Inherit. Metab. Dis._ 13, 301–304 (1990). Article CAS PubMed Google Scholar * Bartholomé, K. & Lüdecke, B. Mutations in the tyrosine
hydroxylase gene cause various forms of L-dopa-responsive dystonia. _Adv. Pharm._ 42, 48–49 (1998). Article Google Scholar * Bonafe, L., Thony, B., Penzien, J. M., Czarnecki, B. &
Blau, N. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. _Am. J. Hum. Genet_ 69,
269–277 (2001). Article CAS PubMed PubMed Central Google Scholar * Vissers, L. E. L. M. et al. A clinical utility study of exome sequencing versus conventional genetic testing in
pediatric neurology. _Genet. Med._ 19, 1055–1063 (2017). Article PubMed PubMed Central Google Scholar * Wright, C. F., FitzPatrick, D. R. & Firth, H. V. Paediatric genomics:
diagnosing rare disease in children. _Nat. Rev. Genet._ 19, 253–268 (2018). Article CAS PubMed Google Scholar * Ng, S. B. et al. Exome sequencing identifies the cause of a mendelian
disorder. _Nat. Genet._ 42, 30–35 (2010). Article CAS PubMed Google Scholar * Ng, S. B. et al. Targeted capture and massively parallel sequencing of 12 human exomes. _Nature_ 461,
272–276 (2009). Article ADS CAS PubMed PubMed Central Google Scholar * Boycott, K. M., Vanstone, M. R., Bulman, D. E. & MacKenzie, A. E. Rare-disease genetics in the era of
next-generation sequencing: discovery to translation. _Nat. Rev. Genet._ 14, 681–691 (2013). Article CAS PubMed Google Scholar * Blau, N., Barnes, I. & Dhondt, J. L. International
database of tetrahydrobiopterin deficiencies. _J. Inherit. Metab. Dis._ 19, 8–14 (1996). Article CAS PubMed Google Scholar * Chabra, S. Clearing the confusion about completed weeks of
gestation. _J. Obstet. Gynecol. Neonatal. Nurs._ 43, 269 (2014). Article PubMed Google Scholar * Spong, C. Y. Defining “term” pregnancy: Recommendations from the defining “term” pregnancy
workgroup. _JAMA_ 309, 2445–2446 (2013). Article CAS PubMed Google Scholar * Leviton, A., Holmes, L. B., Allred, E. N. & Vargas, J. Methodologic issues in epidemiologic studies of
congenital microcephaly. _Early Hum. Dev._ 69, 91–105 (2002). Article PubMed Google Scholar * Sharma, D., Shastri, S. & Sharma, P. Intrauterine growth restriction: antenatal and
postnatal aspects. _Clin. Med. Insights Pediatr._ 10, 67–83 (2016). PubMed PubMed Central Google Scholar * Fenton, T. R. & Kim, J. H. A systematic review and meta-analysis to revise
the Fenton growth chart for preterm infants. _BMC Pediatr._ 13, 59 (2013). Article PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We thank all patients and
their families for their contributions to this study and for their trust. T.H. and J.K. were supported the grant from the Ministry of Health of the Czech Republic RVO-VFN 64165
GJIH-0599-00-7-846 and ProgresQ26/LF1. A.G.C. and N.J.P. are supported by FIS P118/00111 “Instituto de Salud Carlos III (ISCIII)” and “Fondo Europeo de desarrollo regional (FEDER)”. T.O.,
K.J., G.F.H. and O.K.H. were supported in parts by the Dietmar Hopp Foundation, St. Leon-Rot, Germany. M.A.K. is funded by an NIHR Professorship, the Sir Jules Thorn Award for Biomedical
Research and the Rosetrees trust. M.V. is supported by Stichting Stofwisselkracht Grant. D.H. acknowledges funding by the Molecular Diagnostics Program of the National Center for Tumor
Diseases (NCT) Heidelberg. We are grateful for fruitful collaboration with the following clinical partners and patient support groups: Dr. Manolis Bilanakis from the University of Athens,
Aghia Sofia Hospital, Athens, Greece, Prof. Carla Carducci, Dr. Claudia Carducci and Prof. Antonio Angeloni from the Department of Experimental Medicine, Sapienza University of Rome, Italy,
DeNeu and Proyecto Pol, Spain, AADC Research Trust, United Kingdom and The association Lil’ Brave One (Hrabriša), Serbia. We also thank the Society for the Relief of Disabled Children in
Hong Kong for donations to support the commencement of CSF neurotransmitter analyses in Hong Kong. FUNDING Open Access funding enabled and organized by Projekt DEAL. AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * University Children’s Hospital Heidelberg, Division of Child Neurology and Metabolic Disorders, Heidelberg, Germany Oya Kuseyri Hübschmann, Georg F. Hoffmann,
Kathrin Jeltsch, Birgit Assmann & Thomas Opladen * University of British Columbia, Department of Pediatrics, Division of Biochemical Genetics, BC Children’s Hospital, Vancouver, BC,
Canada Gabriella Horvath * Inborn errors of metabolism Unit, Department of Neurology, Institut de Recerca Sant Joan de Déu and CIBERER-ISCIII, Barcelona, Spain Elisenda Cortès-Saladelafont,
Natalia Alexandra Julia Palacios & Angeles García-Cazorla * Inborn Errors of Metabolism and Child Neurology Unit, Department of Pediatrics, Hospital Germans Trias i Pujol, Badalona and
Faculty of Medicine, Universitat Autònoma de Barcelona, Barcelona, Spain Elisenda Cortès-Saladelafont * Hacettepe University, Faculty of Medicine, Department of Pediatrics, Section of
Metabolism, Ankara, Turkey Yılmaz Yıldız & H. Serap Sivri * Department of Human Neuroscience, Unit of Child Neurology and Psychiatry, Università degli Studi di Roma La Sapienza, Rome,
Italy Mario Mastrangelo, Vincenzo Leuzzi & Filippo Manti * First Department of Pediatrics of the University of Athens, Aghia Sofia Hospital, Athens, Greece Roser Pons * UCSD Departments
of Neuroscience and Pediatrics; Rady Children’s Hospital Division of Neurology, Rady Children’s Institute for Genomic Medicine, San Diego, CA, USA Jennifer Friedman * Division of Clinical
and Metabolic Genetics, Department of Pediatrics, University of Toronto, The Hospital for Sick Children 555 University Avenue Toronto, Toronto, ON, Canada Saadet Mercimek-Andrews *
Department of Pediatrics and Adolescent Medicine, The Hong Kong Childrenś Hospital, Hong Kong, Hong Kong Suet-Na Wong & Cheuk-Wing Fung * Department of Neurology, Washington University
School of Medicine, St. Louis, MO, USA Toni S. Pearson * First Department of Pediatrics Aristotle University of Thessaloniki Egnatia St. 106, Thessaloniki, Greece Dimitrios I. Zafeiriou *
Department of Pediatrics and Inherited Metabolic Disorders, First Faculty of Medicine, Charles University and General University Hospital in Prague, Prague, Czech Republic Jan Kulhánek &
Tomáš Honzík * Developmental Neurosciences, UCL Great Ormond Street-Institute of Child Health and Department of Neurology, Great Ormond Street Hospital, London, UK Manju A. Kurian &
Dora Steel * Pediatric Neurology Unit, Department of Pediatrics, University Hospital Reina Sofía, IMIBIC and CIBERER, Córdoba, Spain Eduardo López-Laso & Joaquín Alejandro Fernández
Ramos * Childrenś Department Division of Child Neurology Oslo University Hospital Rikshospitalet Pb 4956 Nydalen, Oslo, Norway Mari Oppebøen * Çukurova University, Faculty of Medicine,
Department of Pediatrics, Division of Pediatric Metabolism and Nutrition, Adana, Turkey Sebile Kılavuz & Halise Neslihan Önenli Mungan * Department of Neurology, Donders Institute for
Brain, Cognition and Behaviour, Radboud University Medical Center, Nijmegen, The Netherlands Tessa Wassenberg * Department of Pediatrics, Pediatric Neurology Unit, UZ Brussel VUB, Brussels,
Belgium Tessa Wassenberg * Department of Pediatrics, University of Alberta Glenrose Rehabilitation Hospital, Edmonton, AB, Canada Helly Goez * Clinic for Pediatrics I, Medical University of
Innsbruck, Innsbruck, Austria Sabine Scholl-Bürgi & Daniela Karall * Department of Pediatrics, AOU Città della Salute e della Scienza, Torino, Italy Francesco Porta * Children’s
Hospital, University Medical Center Hamburg-Eppendorf, Hamburg, Germany René Santer & Philipp Guder * U.O.C. Malattie Metaboliche Ereditarie, Dipartimento della Salute della Donna e del
Bambino, Azienda Ospedaliera Universitaria di Padova – Campus Biomedico Pietro d’Abano, Padova, Italy Alberto Burlina * German Cancer Consortium (DKTK), Heidelberg, Germany Daniel Hübschmann
* Computational Oncology, Molecular Diagnostics Program, National Center for Tumor Diseases, DKFZ, Heidelberg, Germany Daniel Hübschmann * Heidelberg Institute for Stem cell Technology and
Experimental Medicine (HI-STEM), Heidelberg, Germany Daniel Hübschmann * Department of Pediatric Immunology, Hematology and Oncology, Heidelberg University Hospital, Heidelberg, Germany
Daniel Hübschmann * University Children’s Hospital Heidelberg, Dietmar-Hopp Metabolic Center, Heidelberg, Germany Sven F. Garbade * KTP-National University Children’s Medical Institute,
National University Health System, Singapore, Singapore Stacey Tay Kiat Hong * Department of Pediatrics, Showa University School of Medicine, Tokyo, Japan Mitsuhiro Kato * Faculty of
Medicine, University of Novi Sad, Institute for Children and Youth Health Care of Vojvodina, Novi Sad, Serbia Ivana Kavecan * Department of Neurology, Oslo University Hospital, Oslo, Norway
Jeanette Aimee Koht * Department of Neurometabolism and Metabolic Disorders, University Hospital of Nantes, Nantes, France Alice Kuster * University Children’s Hospital, St. Josef-Hospital,
Ruhr-University Bochum, Bochum, Germany Thomas Lücke * Unidad de Trastornos del Movimiento Servicio de Neurología y Neurofisiología Clínica Unidad de Gestión Clínica de Neurociencias
Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocío, Sevilla, Spain Pablo Mir * Department of Pediatrics and Adolescent Medicine, University Medical Centre
Göttingen, Göttingen, Germany Chris Mühlhausen * Clinic of Neurology and Psychiatry for Children and Youth, School of Medicine, University of Belgrade, Belgrade, Serbia Galina Stevanović *
Department of Inborn Errors of Metabolism and Pediatrics, Institute of Mother and Child, Warsaw, Poland Jolanta Sykut-Cegielska * Department of Neurology, Donders Institute for Brain,
Cognition and Behavior, Radboud University Medical Center, Nijmegen, The Netherlands Marcel M. Verbeek * Department of Laboratory Medicine, Translational Metabolic Laboratory (TML), Radboud
University Medical Center, Nijmegen, The Netherlands Marcel M. Verbeek Authors * Oya Kuseyri Hübschmann View author publications You can also search for this author inPubMed Google Scholar *
Gabriella Horvath View author publications You can also search for this author inPubMed Google Scholar * Elisenda Cortès-Saladelafont View author publications You can also search for this
author inPubMed Google Scholar * Yılmaz Yıldız View author publications You can also search for this author inPubMed Google Scholar * Mario Mastrangelo View author publications You can also
search for this author inPubMed Google Scholar * Roser Pons View author publications You can also search for this author inPubMed Google Scholar * Jennifer Friedman View author publications
You can also search for this author inPubMed Google Scholar * Saadet Mercimek-Andrews View author publications You can also search for this author inPubMed Google Scholar * Suet-Na Wong View
author publications You can also search for this author inPubMed Google Scholar * Toni S. Pearson View author publications You can also search for this author inPubMed Google Scholar *
Dimitrios I. Zafeiriou View author publications You can also search for this author inPubMed Google Scholar * Jan Kulhánek View author publications You can also search for this author
inPubMed Google Scholar * Manju A. Kurian View author publications You can also search for this author inPubMed Google Scholar * Eduardo López-Laso View author publications You can also
search for this author inPubMed Google Scholar * Mari Oppebøen View author publications You can also search for this author inPubMed Google Scholar * Sebile Kılavuz View author publications
You can also search for this author inPubMed Google Scholar * Tessa Wassenberg View author publications You can also search for this author inPubMed Google Scholar * Helly Goez View author
publications You can also search for this author inPubMed Google Scholar * Sabine Scholl-Bürgi View author publications You can also search for this author inPubMed Google Scholar *
Francesco Porta View author publications You can also search for this author inPubMed Google Scholar * Tomáš Honzík View author publications You can also search for this author inPubMed
Google Scholar * René Santer View author publications You can also search for this author inPubMed Google Scholar * Alberto Burlina View author publications You can also search for this
author inPubMed Google Scholar * H. Serap Sivri View author publications You can also search for this author inPubMed Google Scholar * Vincenzo Leuzzi View author publications You can also
search for this author inPubMed Google Scholar * Georg F. Hoffmann View author publications You can also search for this author inPubMed Google Scholar * Kathrin Jeltsch View author
publications You can also search for this author inPubMed Google Scholar * Daniel Hübschmann View author publications You can also search for this author inPubMed Google Scholar * Sven F.
Garbade View author publications You can also search for this author inPubMed Google Scholar * Angeles García-Cazorla View author publications You can also search for this author inPubMed
Google Scholar * Thomas Opladen View author publications You can also search for this author inPubMed Google Scholar CONSORTIA INTD REGISTRY STUDY GROUP * Birgit Assmann * , Cheuk-Wing Fung
* , Philipp Guder * , Stacey Tay Kiat Hong * , Daniela Karall * , Mitsuhiro Kato * , Ivana Kavecan * , Jeanette Aimee Koht * , Alice Kuster * , Thomas Lücke * , Filippo Manti * , Pablo Mir *
, Chris Mühlhausen * , Halise Neslihan Önenli Mungan * , Natalia Alexandra Julia Palacios * , Joaquín Alejandro Fernández Ramos * , Dora Steel * , Galina Stevanović * , Jolanta
Sykut-Cegielska * & Marcel M. Verbeek CONTRIBUTIONS O.K.H. and T.O. conceived and designed the study. O.K.H., D.H. and S.F.G. preformed data analysis. O.K.H. drafted the initial
manuscript. O.K.H, S.F.G. and D.H. prepared tables and figures. O.K.H., G.H., T.S.P., S.F.G. and T.O. contributed to finalize manuscript draft with input from all authors. O.K.H., G.H.,
E.C.S., Y.Y., M.M., R.P., J.F., S.M.A., S.W., T.S.P., D.I.Z., J.K., M.A.K., E.L.L., M.O., S.K., T.W., H.G., S.S.B., F.P., T.H., R.S., A.B., H. S.S., V.L., G.F.H., K.J., A.G.C., T.O., B.A.,
C.W.F., P.G., S.T.K.H., D.K., M.K., I.K., J.A.K., A.K., T.L., F.M., P.M., C.M., H.N.Ö.M., N.A.J.P., J.A.F.R., D.S., G.S., J.S.C., M.M.V. contributed to data and patient enrollment. All
authors approved the final version of the manuscript. CORRESPONDING AUTHOR Correspondence to Thomas Opladen. ETHICS DECLARATIONS COMPETING INTERESTS A.G.C. receives teaching honorarium from
PTC Therapeutics GT, Inc. C.M. and E.L.L. have received consultancy honorarium as part of the Advisory Board of PTC Therapeutics GT, Inc. G.F.H. receives teaching as well as consultancy
honorarium from PTC Therapeutics GT, Inc. J.F. had trials with Biogen (Angelman’s Syndrome) and Stealth Biotherapeutics (Mitochondrial Disorders); J.F.’s spouse is Founder and Principal of
Friedman Bioventure, which holds a variety of publicly traded and private biotechnology interests. O.K.H. and T.W. have received teaching honorarium from PTC Therapeutics GT, Inc. R.P. has
received honoraria as a speaker from Genesis Pharma, PTC Therapeutics GT, Inc. and as consultant in Advisory board of PTC Therapeutics GT, Inc. T.O. receives teaching honorarium and research
support from PTC Therapeutics GT, Inc. V.L. has received consultancy honoraria as part of Advisory Boards organized by PTC Therapeutics GT, Inc., BioMarin Pharmaceutical Inc. and Homology
Medicines. The remaining authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks Phillip Pearl and the other anonymous
reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims
in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN
ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format,
as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third
party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the
article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright
holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kuseyri Hübschmann, O., Horvath, G.,
Cortès-Saladelafont, E. _et al._ Insights into the expanding phenotypic spectrum of inherited disorders of biogenic amines. _Nat Commun_ 12, 5529 (2021).
https://doi.org/10.1038/s41467-021-25515-5 Download citation * Received: 09 December 2020 * Accepted: 12 August 2021 * Published: 20 September 2021 * DOI:
https://doi.org/10.1038/s41467-021-25515-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative