Engineering avidcars for combinatorial antigen recognition and reversible control of car function
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT T cells engineered to express chimeric antigen receptors (CAR-T cells) have shown impressive clinical efficacy in the treatment of B cell malignancies. However, the development of
CAR-T cell therapies for solid tumors is hampered by the lack of truly tumor-specific antigens and poor control over T cell activity. Here we present an avidity-controlled CAR (AvidCAR)
platform with inducible and logic control functions. The key is the combination of (i) an improved CAR design which enables controlled CAR dimerization and (ii) a significant reduction of
antigen-binding affinities to introduce dependence on bivalent interaction, i.e. avidity. The potential and versatility of the AvidCAR platform is exemplified by designing ON-switch CARs,
which can be regulated with a clinically applied drug, and AND-gate CARs specifically recognizing combinations of two antigens. Thus, we expect that AvidCARs will be a highly valuable
platform for the development of controllable CAR therapies with improved tumor specificity. SIMILAR CONTENT BEING VIEWED BY OTHERS DIRECT CONTROL OF CAR T CELLS THROUGH SMALL
MOLECULE-REGULATED ANTIBODIES Article Open access 29 January 2021 TUNABLE CONTROL OF CAR T CELL ACTIVITY THROUGH TETRACYCLINE MEDIATED DISRUPTION OF PROTEIN–PROTEIN INTERACTION Article Open
access 09 November 2021 SPLIT-DESIGN APPROACH ENHANCES THE THERAPEUTIC EFFICACY OF LIGAND-BASED CAR-T CELLS AGAINST MULTIPLE B-CELL MALIGNANCIES Article Open access 11 November 2024
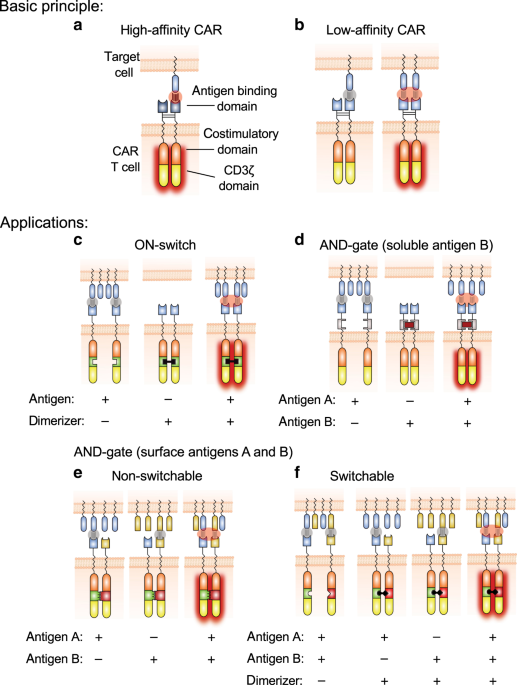
INTRODUCTION CARs are synthetic proteins consisting of antigen-binding domains linked to T-cell-activating signaling domains (Fig. 1a)1. T cells genetically engineered to express CARs (CAR-T
cells) efficiently eliminate antigen-expressing target cells, which is reflected by the impressive efficacy of CD19-directed CAR-T cell therapies in patients with B cell malignancies2. A
major challenge in translating this success to other malignancies is the fact that CAR antigens are almost exclusively tumor-associated antigens (TAAs), which are not truly tumor specific.
Since most TAAs are also expressed in vital healthy tissues, efficient TAA targeting is expected to result in unacceptable on-target/off-tumor toxicity, which has indeed been observed in
several clinical studies3,4,5,6,7. Ultimately, this could preclude increasing the efficacy of CAR-T cell therapies to levels required for sustained remission of solid tumors. The problem of
on-target/off-tumor toxicity is further aggravated by the fact that the activity of CAR-T cells is difficult to control in a reversible manner. To address the lack of control and tumor
specificity associated with CAR-T cells, several promising strategies have been developed including CARs for combinatorial antigen recognition8,9,10,11,12,13,14, affinity-tuned CARs to
minimize killing of healthy cells expressing low levels of target antigen15,16,17,18,19,20 and CARs whose function can be regulated by administration of small molecules or soluble
proteins10,21,22,23,24,25,26,27,28,29. Despite these important advances, there is still a need for improvement. For example, currently available systems for the control of CAR-T cells are
either based on small molecules which are suboptimal for clinical application or on suicide switches which irreversibly destroy the CAR-T cells. Furthermore, previously described AND-gate
CARs suffer from low specificity12,13,14 or their inability to differentiate between a double-positive cell and two single-positive cells, which express the antigens A and B complementarily
and are located in close proximity to each other21,30. That is, in this SynNotch strategy, expression of the CAR can be induced by healthy cells expressing antigen A, enabling recognition of
nearby healthy cells expressing antigen B. Furthermore, the SynNotch system is based on xenogeneic and thus potentially immunogenic proteins. One critical parameter defining the potency and
function of CARs is the affinity of their antigen-binding domains. Importantly, the interaction with a target antigen not only depends on the affinity of an individual antigen-binding
domain, but also on the valency of the interaction. That is, a multivalent interaction amplifies the individual affinities through a phenomenon known as avidity. Many types of natural immune
reactions are based on low affinities, which are amplified by avidity effects, such as antigen binding by IgM during early humoral immune responses. However, the influence of avidity
effects on CAR function has mostly been neglected in the CAR field so far, even though most currently used CARs are based on naturally dimeric components31,32,33. Here, we demonstrate that
this aspect can be exploited for the integration of inducible and logic control functions into CAR molecules. We show that it is possible to generate avidity-controlled CARs (AvidCARs) which
are highly potent and dependent on bivalent antigen engagement (Fig. 1). These AvidCARs are based on two main design principles: controlled CAR dimerization and low-affinity antigen
binding. We exemplify the potential of the AvidCAR platform (1) by generating a controllable ON-switch CAR that can be regulated by CAR homodimerization with a clinically applied small
molecule (Fig. 1c); (2) by constructing an AND-gate CAR that is triggered only in the presence of both a surface molecule on tumor cells and a soluble protein found in tumor stroma (Fig.
1d); and (3) by generating a heterodimeric AND-gate CAR which enables the combinatorial recognition of two different antigens exclusively when co-presented on the surface of the same cell.
This latter AND-gate AvidCAR is designed either in a constitutive (Fig. 1e) or in a switchable version regulated by a dimerizing small molecule (Fig. 1f). Finally, we also investigate the
suitability of different antigen-binding modules and various co-stimulatory domains for the design of efficient AvidCARs. Together, our study illustrates the potential of exploiting avidity
effects and demonstrates that the AvidCAR platform is a versatile concept for inducible and combinatorial CAR control. RESULTS CONSTRUCTION OF AVIDITY-CONTROLLED CARS Since a critical design
principle of AvidCARs is the control over CAR dimerization, the usage of CAR components potentially causing uncontrolled dimerization or oligomerization needs to be avoided. Given the
recent report on scFv-mediated CAR tonic signaling (probably caused by scFv-mediated clustering)34, we looked for an antigen-binding domain based on a single protein domain, which prevents
intermolecular dimerization as was observed with several scFvs35,36,37. Therefore, we chose the human epidermal growth factor receptor (hEGFR)-specific binder E11.4.1, which was previously
engineered based on the single-domain protein rcSso7d38. As described above, we hypothesized that the construction of an AvidCAR also requires the incorporation of low-affinity binding
domains (Fig. 1b). For this purpose, we reduced the affinity of the high-affinity binder E11.4.1 by performing an alanine scan, resulting in eight different E11.4.1 mutants. To determine the
affinities of those mutants, they were expressed as monomeric proteins (Supplementary Fig. 1a) and titrated on Jurkat cells expressing truncated hEGFR (hEGFRt, Supplementary Fig. 1c). These
titration experiments yielded an affinity of 13 nM for the high-affinity binder E11.4.1, which is in close agreement with the previously published value (17 nM)38. Among the mutants we
identified two (E11.4.1-G25A and E11.4.1-G32A) with substantially decreased, yet measurable affinities of 125 and 877 nM, respectively (Fig. 2a). Complementary affinity determination by
surface plasmon resonance (SPR) analysis with the entire extracellular domain of hEGFR yielded highly comparable _K_d values (Supplementary Fig. 1b). To test our hypothesis that only
low-affinity, but not high-affinity CARs are dependent on bivalent interaction, we incorporated the high-affinity (E11.4.1; _K_d of 13 nM), intermediate-affinity (E11.4.1-G25A; _K_d of 125
nM), or low-affinity (E11.4.1-G32A; _K_d of 877 nM) hEGFR binders into CARs comprising the hinge and transmembrane region of CD8α, followed by the signaling domains of 4-1BB and CD3ζ (termed
BBz-CAR, Fig. 2b). In addition, each of those three CARs was also created in a second version in which the two extracellular cysteine residues in the CD8α hinge region, which are known to
cause CD8α dimerization32, were mutated to serine residues, resulting in a CAR backbone termed Ser-BBz. All CAR variants were expressed at similar levels in primary human T cells
(Supplementary Fig. 2a). In co-culture with hEGFRt-expressing target cells (Supplementary Fig. 2b) both the high- and intermediate-affinity CARs triggered target cell lysis and IFN-γ
secretion both in the BBz-, as well as in the Ser-BBz-format, albeit at slightly higher efficiency in the dimeric BBz version (Fig. 2c and Supplementary Fig. 2c). In contrast, the
low-affinity CAR only mediated lysis and IFN-γ production when expressed with dimerization-promoting cysteine residues in its CD8α hinge (Fig. 2c and Supplementary Fig. 2c), thus supporting
our initial hypothesis that low-affinity CARs are dependent on bivalent interactions. Next, we used a mathematical model to investigate whether the observed avidity-driven activation of
AvidCARs is mediated by an increase in the effective lifetime of the CAR–antigen interaction (by allowing for increased rebinding) and/or by increased binding (i.e., more CAR occupancy by
antigen). A longer lifetime is thought crucial for pMHC binding to the T-cell receptor (TCR) as a result of kinetic proofreading and this has recently been shown to be the case also for
CARs39. Thus, to investigate how avidity alters CAR occupancy and lifetime, we set up a mathematical model of antigen binding to a bivalent CAR with kinetic proofreading (e.g., representing
CAR phosphorylation) where a tunable parameter (_σ_) was used to modulate avidity (Supplementary Fig. 3a). The model shows that increasing avidity has only a modest effect on CAR occupancy
but a dramatic effect on the amount of phosphorylated CAR, highlighting that increased avidity strongly prolongs the duration of individual CAR–antigen interactions, thereby enhancing the
signaling capacity. In summary, this mathematical model predicts that increased avidity leads to improved antigen sensitivity (Supplementary Fig. 3b). To investigate this dependence of
AvidCAR sensitivity on avidity experimentally, we switched to another assay system based on small molecule-mediated CAR dimerization. For that purpose, the FK506-binding protein (FKBP)12 and
a mutant FKBP12-rapamycin binding domain (FRB) were integrated into the Ser-BBz-CARs for small molecule (AP21967)-induced CAR heterodimerization (Fig. 2d). To additionally investigate the
impact of antigen dimerization on avidity, we integrated the FKBP12-F36V domain into hEGFRt for small molecule (AP20187)-mediated antigen homodimerization (Fig. 2d). Primary human T cells
expressing either high-affinity CARs or the corresponding low-affinity AvidCARs were then co-cultured ± AP21967 (for CAR dimerization) and ± AP20187 (for antigen dimerization) with Jurkat
target cells expressing the antigen hEGFRt at different densities ranging from hundreds to several hundred thousand molecules per cell (Supplementary Fig. 4a). Target cell lysis mediated by
the high-affinity CAR was largely independent of the dimerization state of both the CAR and the antigen, strongly suggesting that the high-affinity CAR was not dependent on bivalent
interaction (Fig. 2e, left diagram). In contrast, the low-affinity AvidCAR efficiently triggered activation only in a dimeric form (Fig. 2e, right diagram, black and red solid lines), again
demonstrating its dependency on bivalent interaction, i.e., avidity. Furthermore, the antigen sensitivity of this dimeric AvidCAR was strongly increased if also the antigen was dimerized
(red solid line vs. black solid line, Fig. 2e, right diagram). Thus, enhancing the avidity effect of the dimeric AvidCAR—mediated through additional antigen dimerization in this
experiment—resulted in increased antigen sensitivity, as predicted by the mathematical model described above. Similar activation patterns were observed when measuring IFN-γ release
(Supplementary Fig. 4b). High-affinity CAR and AvidCAR constructs were expressed at similar levels, hence excluding expression-based effects (Supplementary Fig. 4c). Together, these
experiments demonstrate that it is possible to engineer AvidCARs which depend on bivalent interaction and whose function can thus be regulated by drug-mediated dimerization. Moreover, the
comparable efficacy of the monomeric and dimeric high-affinity CAR (Fig. 2e) is in line with a previous report showing that a single CD3ζ domain is sufficient for efficient signaling40.
SCFVS MAY PRECLUDE CONTROLLED CAR DIMERIZATION Global efforts in the development and clinical testing of novel CAR-T cell constructs almost exclusively rely on scFv-based binders. To test
the suitability of scFvs for generating AvidCARs, we incorporated the hHER2-specific low-affinity scFv 4D5-5 (_K_d of 1.1 µM)15 into both the BBz- and the Ser-BBz-CAR (Fig. 3a). In contrast
to the rcSso7d-based CARs described above, in the scFv-based CAR the Cys→Ser-mutation had little effect on target cell lysis and IFN-γ secretion by CAR-T cells co-cultured with hHER2tpos
target cells (Fig. 3b), despite even lower expression levels of the CAR containing the Ser-mutation (Supplementary Fig. 5a). Given those functional differences compared to the rcSso7d-based
CARs despite similarly low affinities, we hypothesized that scFv-mediated oligomerization might prevent the formation of strictly monomeric CARs. Dimerization of scFvs has previously been
reported for soluble scFvs, resulting in the formation of so-called diabodies, in which the VH of one scFv molecule pairs with the VL of another scFv molecule35,36,37 (Fig. 3c). To
investigate whether intermolecular dimerization of VH and VL between scFvs can also trigger CAR oligomerization, we expressed this scFv as an Fv, i.e., without the linker connecting VL and
VH, thus preventing such intermolecular dimerization (Fig. 3d and Supplementary Fig. 5b). In this Fv-based CAR, the VH was fused to the Ser-BBz-backbone, whereas the VL was anchored via a
separate CD8α(Ser) transmembrane domain without an intracellular signaling region. Importantly, compared to the scFv-based CAR, significantly reduced cytotoxicity was observed with the
Fv-based CAR (Fig. 3e), strongly suggesting that removing the linker from the scFv prevents CAR dimerization and thereby avidity-based activation of low-affinity CARs. Of note, CAR activity
could be restored by AP20187-induced homodimerization of two Fv-based CAR molecules (Fig. 3e). This demonstrates (1) that separate VH- and VL-containing constructs assemble into a CAR
molecule with a functional Fv unit and (2) that in the absence of scFv-mediated oligomerization, avidity can be controlled by inducing dimerization. As expected, in the absence of the VL
construct the dimerization of the VH-construct had no effect, confirming that antigen binding requires an assembled Fv comprising VH and VL (Fig. 3e). Furthermore, all constructs were
expressed at similar levels, thus excluding expression-mediated effects (Supplementary Fig. 5c). Together, these data demonstrate that intermolecular VH/VL dimerization of at least certain
scFvs can cause CAR oligomerization. As a consequence, such clustering scFvs are undesired in our context not only because of tonic CAR signaling34,41, but also because of preventing the
avidity-based control of AvidCARs. INFLUENCE OF DIFFERENT CO-STIMULATORY DOMAINS Next, we investigated whether the dependence of low-affinity CARs on dimerization is also observed with other
co-stimulatory domains by exchanging the 4-1BB domain in the Ser-BBz-CAR backbone for the endodomain of CD28, ICOS, OX40, or CD2 in our hEGFR-specific rcSso7d-based AvidCAR as described
above. All CARs additionally contained an FKBP12-F36V domain for conditional homodimerization by AP20187 (Fig. 3f). In a cytotoxicity experiment with primary human T cells expressing those
CARs, significant dimerization dependence was observed with 4-1BB-, ICOS- and CD2-mediated co-stimulation, but not if CD28 or OX40 were incorporated (Fig. 3g). IFN-γ secretion was
significantly induced by dimerization of 4-1BB- and CD2-based CARs (Supplementary Fig. 5e). Albeit not being significant, a trend was also observed with the endodomain of OX40. In summary,
these data suggest that among the co-stimulatory domains tested, the ones derived from 4-1BB or CD2 are best suited for construction of efficient AvidCARs—at least with the binding
affinities and antigen densities tested in these experiments. AVIDCARS WITH AND-GATE FUNCTION Current bispecific CARs are characterized by high-affinity binding domains and, hence, do not
possess AND-gate function but OR-gate function, i.e., activation in the presence of either antigen A or B or both42,43,44. Moreover, the homodimeric/-oligomeric nature of current CAR designs
facilitates multivalent engagement of a single antigen by several identical binding domains, thus additionally precluding efficient AND-gate function. Here, we tested whether our AvidCAR
platform, in which undesired homodimerization can be prevented, allows for the generation of bispecific CARs with robust AND-gate function. To this end, we generated an additional
low-affinity binder recognizing a different model antigen. As a starting point, we chose the hHER2-specific single-domain binder zHER2-AK, which is based on the previously engineered
affibody ZHER2:445. High-affinity binding of zHER2-AK to hHER2 was confirmed by SPR, yielding a _K_d of 17 nM (Supplementary Fig. 6b). Again, to reduce its affinity, the affibody was
subjected to an alanine scan. The resulting 12 mutants were incorporated into the Ser-BBz-CAR backbone comprising the FKBP12-F36V domain for functional screening by controlled
homodimerization. In a cytotoxicity assay, the CAR containing the binder mutant zHER2-AK-R10A triggered lysis of hHER2tpos cells in a dimerization-dependent manner (Supplementary Fig. 6a),
indicating that the reduced affinity of this mutant necessitates bivalent antigen engagement. Indeed, subsequent SPR analysis confirmed that this affibody-mutant binds to hHER2 with low
affinity (_K_d of 865 nM), which is comparable to that of the hEGFR-specific rcSso7d-based binder E11.4.1-G32A described above. To generate the hEGFR/hHER2-bispecific AND-gate AvidCAR, those
two low-affinity binders were incorporated into two separate Ser-BBz-CAR constructs with complementary heterodimerization domains (Fig. 4a). As we had hypothesized, primary human T cells
expressing this AND-gate AvidCAR efficiently triggered lysis of target cells expressing both hEGFRt and hHER2t, while lysis of cells expressing only hEGFRt or hHER2t was comparable to
background lysis by CARneg T cells (Fig. 4b and Supplementary Fig. 6c). This experiment clearly demonstrates the dependency of this heterodimeric AvidCAR on combinatorial antigen
recognition. Moreover, cytotoxicity was only observed in the presence of the heterodimerizing agent, demonstrating that this AND-gate AvidCAR can be additionally controlled by administration
of a small molecule. Of note, the incorporation of the high-affinity versions of both binders (_K_d values of 13 and 17 nM for E11.4.1 and zHER2-AK, respectively) abrogated the AND-gate
character and the dependence on dimerization (Fig. 4b), thus confirming the necessity of low-affinity antigen binding. Both the high-affinity and the low-affinity heterodimeric CARs were
expressed at similar levels, excluding any major expression-based bias (Supplementary Fig. 6d). Fusion of the two low-affinity binders in tandem into a single, monomeric CAR molecule
(TanCAR) (Fig. 4a), also yielded specificity for double-positive target cells (Fig. 4b), albeit without the possibility to conditionally control CAR function with a small molecule. Together,
these data demonstrate that bispecific AvidCARs can be engineered for robust combinatorial antigen recognition, which ultimately allows for specific elimination of target cells
co-expressing two antigens on their surface, but not their single-positive counterparts. AND-GATE WITH A SURFACE AND A SOLUBLE ANTIGEN Based on the data described above, we hypothesized that
the AvidCAR concept also enables the construction of CARs that are specifically activated by combinatorial recognition of a surface antigen and a tumor-resident soluble factor, which in
this case acts as a CAR-dimerizing agent. For that purpose, we designed a hEGFR-specific AvidCAR additionally containing the heterodimeric Fc antigen binding (Fcab) mutant JanusCT6, which
recognizes vascular endothelial growth factor (VEGF)46 and expressed this AvidCAR construct in primary human T cells (Supplementary Fig. 6e). As VEGF is naturally present as a dimer, we
hypothesized that it could homodimerize the AvidCAR molecules and thereby facilitate bivalent recognition of hEGFR (Fig. 4c). Indeed, VEGF triggered lysis of hEGFRt-expressing Jurkat cells,
thus confirming our hypothesis (Fig. 4d). Of note, VEGF had no effect on target cell viability in the presence of T cells expressing either no CAR or a VEGF-independent high-affinity CAR,
thus excluding any direct toxic effects caused by VEGF. Summing up, we designed an AvidCAR that recognizes the combination of a cell surface and a soluble antigen, thus further broadening
the application range of the AvidCAR platform. REGULATION AND SPECIFICITY OF AVIDCARS IN VIVO As shown above, the activity of ON-switch AvidCARs can be regulated by conditional dimerization
with small molecules. To test this principle in vivo, we lentivirally transduced primary human T cells with a hEGFR-specific ON-switch AvidCAR, whose antigen-binding domain specifically
recognizes human, but not murine EGFR (mEGFR) (Supplementary Fig. 7a). This CAR additionally harbors the FKBP12-F36V domain for conditional homodimerization by AP20187. CD19pos Nalm-6 target
cells were stably transduced with hEGFRt (Supplementary Fig. 7b), enabling the direct comparison of the hEGFR-directed AvidCAR with two control CARs: (1) a high-affinity version of this
AvidCAR and (2) a CD19-specific reference BBz-CAR based on the scFv FMC63 (αCD19-BBz; Supplementary Data 1). Initial in vitro experiments confirmed that (1) both control CARs (αCD19-BBz-CAR
and high-affinity αhEGFR-Ser-BBz-CAR) efficiently triggered lysis of CD19pos/hEGFRtpos Nalm-6 cells, (2) that the activity of the hEGFR-specific ON-switch AvidCAR was dependent on the
presence of the dimerizer AP20187 (Fig. 5a) and that (3) the parental hEGFRtneg Nalm-6 cells were not recognized by hEGFR-directed CARs (Fig. 5b). Similar effects were observed irrespective
of the effector:target ratio used (Fig. 5a and b). In vivo, both control CARs efficiently delayed tumor outgrowth. Remarkably, the switchable αhEGFR-AvidCAR mediated comparable tumor
control, but only upon administration of the dimerizing small molecule (on-state; Fig. 5c, Supplementary Fig. 7c). Moreover, even if the switchable αhEGFR-AvidCAR-T cells were initially
administered without the dimerizing small molecule (off-state, leading to tumor outgrowth), the data indicate that antitumor activity could still be triggered by a single administration of
the small molecule 11 days after CAR-T cell administration (Fig. 5c and Supplementary Figs. 7c and 8a). To investigate the effects of these CAR-T cells over a longer time span, we generated
a Kaplan–Meier curve (Fig. 5d). Again, while the αhEGFR-AvidCAR-T cells had no effect in the absence of the dimerizer, long-term administration of the dimerizing small molecule resulted in
significant prolongation of survival—similar to that observed upon treatment with either the high-affinity version of the αhEGFR-CAR or the standard αCD19-BBz-CAR. Together, these results
confirm that CAR function can be controlled in vivo by regulating CAR avidity. Importantly, this AvidCAR represents a clinically applicable ON-switch CAR, as the domain FKBP12-F36V (used for
conditional homodimerization by AP20187 in this experiment) can also be homodimerized by the closely related investigational drug AP1903 (rimiducid). Finally, we also tested the AND-gate
function of an AvidCAR in vivo. For this purpose, we inserted domains for constitutive heterodimerization into the hEGFR/hHER2-specific AvidCAR described above, since the persistent
administration of sufficient amounts of the currently available heterodimerizer AP21967 in mice is not feasible21. That is, the domains for conditional heterodimerization were replaced with
drug-independent heterodimerizing leucine zipper domains10,47 (EE and RR, respectively; Fig. 6a; Supplementary Data 1). The two polypeptide chains of this αhEGFR/αhHER2-AvidCAR were
expressed at high levels (Supplementary Fig. 8b,c) and formed a functional CAR with potent AND-gate function, as demonstrated by specific lysis of hEGFRtpos/hHER2tpos target cells, but no or
only weak lysis of single-positive target cells (Fig. 6b). Subsequently, this αhEGFR/αhHER2-AvidCAR was introduced lentivirally into primary human T cells and tested for its function in an
in vivo model, where four different Nalm-6 populations (hEGFRt/hHER2t double-positive Nalm-6, both types of single-positive controls and double-negative version) were mixed 1:1:1:1 and
subsequently administered intravenously (i.v.) to NSG mice. Ten days after CAR-T cell inoculation, the four different Nalm-6 populations were analyzed in the bone marrow, liver, blood, and
spleen (Fig. 6c, d, and Supplementary Figs. 9–11). Strikingly, the αhEGFR/αhHER2-AvidCAR-T cells specifically killed double-positive, while ignoring single-positive Nalm-6 cells, even though
they were present in the same tissue of the same animal (Fig. 6c, d and Supplementary Fig. 10). Notably, this high specificity was obtained despite even higher antigen expression levels in
the single-positive controls (Fig. 6c and Supplementary Fig. 8d). In addition to its high specificity, the efficacy of the αhEGFR/αhHER2-AvidCAR-T cells was comparable to that of
high-affinity αhEGFR-CAR-T cells (equally efficient in the liver; only slightly less efficient in the bone marrow; Fig. 6d and Supplementary Fig. 10). No Nalm-6 cells were detected in the
blood and spleen of diseased mice (Supplementary Fig. 11). As expected, the high-affinity αhEGFR-CAR mediated killing of both hEGFRt/hHER2t double-positive and hEGFRt single-positive Nalm-6
cells (Fig. 6c, d and Supplementary Fig. 10). Together, these in vivo data demonstrate that the αhEGFR/αhHER2-AvidCAR is both efficacious and highly specific, even if double-positive target
cells and single-positive controls are co-localized in the same tissues. To the best of our knowledge, this is the first example of an AND-gate CAR that can selectively target
double-positive cells without requiring a safe distance between single-positive healthy bystander cells. DISCUSSION In this study we present the concept of AvidCARs. The key design
principles were (1) the development of strictly monomeric CAR backbones by removing cysteines in the hinge region to prevent uncontrolled disulfide-mediated CAR dimerization; (2) the
incorporation of antigen-binding domains whose affinities are substantially reduced down to levels comparable to those of TCRs (_K_d > 0.5 µM)48; and (3) the use of single-domain
antigen-binding domains, which seems to be preferable to exclude potential oligomerization that has been observed for some scFvs. As described above, AvidCARs were successfully engineered
using an EGFR-targeting rcSso7d-based binder and an affibody directed against HER2. Of note, similar dimerization-dependent AvidCAR activation was also observed when using low-affinity
nanobodies targeting green fluorescent protein (GFP)49 and HER250, (Supplementary Fig. 12). Thus, in total we have successfully generated AvidCARs with four different targeting domains based
on three different protein scaffolds, demonstrating the generalizability of the AvidCAR concept. An important feature, that all of these targeting domains (rcSso7d, affibodies and
nanobodies) have in common, is their single-domain architecture. In contrast, scFvs are composed of two domains, which are known to cause mispairing and thus oligomerization to various
extent depending on the scFv mutant, linker length, and environmental conditions51,52. This has long been known for soluble scFvs52 and recently such scFv clustering has also been associated
with tonic signaling in CAR-T cells34. In our study, we provide direct biochemical evidence that scFv clustering indeed takes place on the CAR-T cell surface. Moreover, our experiments also
demonstrated that scFvs that tend to cluster do not allow for the generation of efficient AvidCARs. However, we do not exclude that other scFvs may be suitable for the construction of
AvidCARs. We would also like to note that we do not propose the separate expression of VH and VL as a suitable solution in case of oligomerizing scFvs. This design was only used as a model
system to study the mechanism of scFv clustering (Fig. 3d, e). Instead, we strongly favor the use of single-domain binding scaffolds, especially in the context of AvidCARs. In fact, various
different single-domain binding scaffolds have already been used in CARs and due to their favorable properties might be attractive for CARs in general. Regarding the potential immunogenicity
it should be noted that while the single-domain binding scaffolds used here are based on nonhuman proteins, there are also several alternatives derived from human protein domains, such as
monobodies. Our experiments demonstrate that AvidCARs with low-affinity antigen-binding domains require bivalent interaction to be efficiently triggered. Such behavior can be explained by
the fact that CAR signaling, i.e., the sufficient phosphorylation of signaling proteins, requires a certain duration of interaction with its target antigens, as has been shown for the TCR
and other immunoreceptors39,53,54,55. For low-affinity binding domains, this duration is too short in a monovalent interaction and must be prolonged by interaction via a second binding
domain, as supported by both our mathematical model and experimental data, in particular those presented in Fig. 2e. We demonstrate that this mechanism can be exploited to regulate CAR
function by conditional dimerization and/or for combinatorial recognition of two different antigens. Such AND-gate function mediated by bivalent interaction of a bispecific CAR would not be
possible with current homodimeric CAR formats, which may explain why bispecific CARs have so far only been used as OR gates42,43,44. Finally, due to the dependence on bivalent interaction,
we could also use our AvidCAR platform as a functional reporter system for identifying clustering tendencies of different CAR components such as a hHER2-specific scFv. The AND-gate AvidCAR
presented in Fig. 4a, b combines both small molecule-mediated regulation (ON-switch function) and combinatorial antigen recognition (AND-gate function). However, since the required small
molecule AP21967 is not suited for long term in vivo experiments21, we replaced this AP21967-responsive heterodimerization module with a constitutive version based on leucine zippers to be
able to study this AND-gate AvidCAR in vivo (Fig. 6). Of note, recently we introduced a small molecule-regulated heterodimerization system, which is based on human retinol binding protein 4,
an engineered protein scaffold and the orally available small-molecule drug A112029. Thus, this molecular ON-switch represents an alternative to the AP21967-based system, potentially
enabling the future design of AND-gate AvidCARs that can additionally be controlled with a small-molecule drug shown to be nontoxic in mice, even when administered at high doses up to 30
mg/kg for several months56. A crucial feature of the avidity-based AND-gate function of AvidCARs is the fact that antigens A and B must be co-expressed on the same cell. This is in contrast
to SynNotch CAR-T cells, in which CAR expression can be induced by healthy cells expressing antigen A, which subsequently enables the recognition of nearby healthy cells expressing antigen
B30. Another potential advantage of AvidCAR-T cells is that they are also fully functional when target cells are rare, as AvidCAR expression—unlike CAR expression in SynNotch T cells—does
not need to be induced by antigen-expressing cells. Finally, the independence of AvidCARs from a xenogeneic transcription factor reduces the risk of immunogenicity. Another consequence of
the avidity-based mechanism of AvidCARs is the strong capability to sense antigen density. Antigen density determines the probability that a second binding domain can catch an antigen before
the first one dissociates from its antigen. That is, antigen density strongly determines the lifetime of the interaction between a bivalent low-affinity CAR and its antigens—and thus CAR
signaling. This mechanism is exploited in affinity tuning of current CARs to limit CAR-T cell cytotoxicity to tumor cells expressing elevated target antigen levels15,16,17,18,19,20. Due to
the same mechanism, AND-gate AvidCARs integrate the dependence on two different antigens (i.e., AND-gate function) with the ability to sense antigen density, which can further increase tumor
specificity. Nevertheless, for other applications, it is desirable to enhance the antigen sensitivity of the AvidCARs, e.g., for antigens expressed at low levels and/or to reduce the risk
of tumor escape due to antigen downregulation. It is expected that avidity effects—and thereby CAR sensitivity—can be further increased by optimizing the design of AvidCARs (e.g., length and
flexibility of spacers), which was not investigated in the examples presented here. However, we show that antigen dimerization enhances the avidity effect and thereby AvidCAR sensitivity.
Therefore, co-localized antigens are particularly attractive candidates. Of note, many TAAs naturally exist in a dimeric/oligomeric state57,58,59, are clustered into nanodomains or interact
directly with other antigens. Prominent examples of co-localization include heterodimers of EGFR family members either among themselves or with other growth factor receptors, or the
co-localization of receptors and gangliosides60,61,62,63. The architecture of the mammalian cell membrane is still incompletely understood and many more co-localized proteins may exist that
are not known yet64,65. Together, our study highlights and explores a previously neglected aspect in the CAR field, which is the bivalent antigen engagement due to the dimeric nature of
virtually all currently applied CAR molecules. Therefore, the presented data will contribute to an improved mechanistic understanding of CAR function and biology. Moreover, we present a
strategy for specifically exploiting this bivalent antigen recognition in a controlled manner, ultimately opening up a range of applications in which an AvidCAR integrates multiple input
signals, e.g., (1) a cell surface antigen and a clinically applied small molecule, (2) two different cell surface antigens and a small molecule or (3) a cell surface antigen and a soluble
factor enriched in the tumor microenvironment. Thus, we expect that AvidCARs will be a highly valuable platform for the development of controllable CAR therapies with improved tumor
specificity. METHODS CELL CULTURE Buffy coats from de-identified healthy donor’s blood were purchased from the Austrian Red Cross, Vienna, Austria. Primary human T cells were isolated by
negative selection using the RosetteSep Human T-Cell Enrichment Cocktail (STEMCELL Technologies) and cryopreserved in RPMI-1640 GlutaMAX medium (Thermo Scientific) supplemented with 20% FCS
(Sigma-Aldrich) and 10% DMSO (Sigma-Aldrich) until further use. After thawing, primary human T cells were immediately activated with Dynabeads Human T-Activator αCD3/αCD28 beads (Thermo
Scientific) at a 1:1 ratio according to the manufacturer’s instructions. T cells were expanded before the experiments for at least 14 days in T-cell medium, consisting of RPMI-1640 GlutaMAX
supplemented with 10% FCS, 1% penicillin–streptomycin (Thermo Scientific) and 200 U/mL recombinant human IL-2 (Peprotech). Half of the medium was exchanged every other day and cell densities
were kept between 0.4 and 2 × 106 cells/mL. Nalm-6 cells and Jurkat cells (gifts from Dr. Sabine Strehl and Dr. Michael Dworzak, respectively; CCRI, Vienna, Austria) were maintained in
RPMI-1640 GlutaMAX supplemented with 10% FCS and 1% penicillin–streptomycin. Lenti-X 293T cells (Takara) were maintained in DMEM (Thermo Scientific) supplemented with 10% FCS. Nalm-6 target
cell lines ± hEGFRt and/or ± hHER2t expression were established by transduction with a lentiviral vector encoding firefly luciferase (ffLuc), puromycin N-acetyl transferase and hEGFRt and/or
hHER2t. A stable clone of Nalm-6 cells expressing both transgenes was established by limiting dilution cloning and subsequently was co-transduced with a lentiviral vector encoding ffLuc and
enhanced GFP (eGFP) to ensure high expression of the luciferase reporter gene. Cell lines were regularly tested for mycoplasma contamination using the MycoAlert PLUS Mycoplasma Detection
Kit (Lonza) and for squirrel monkey retrovirus contamination by PCR (performed by Labdia Labordiagnostik GmbH, Austria). Cell line authentication was performed by SNP-profiling at
Multiplexion GmbH, Germany. The homo- and heterodimerization of proteins in vitro was induced by the addition of AP20187 or AP21967 (both Takara) to the cell suspensions at a final
concentration of 10 and 500 nM, respectively. The effective concentrations of both compounds were determined by the addition of increasing concentrations to Jurkat cells expressing hEGFRt,
which could be homo- or heterodimerized (Supplementary Fig. 4d). IN VITRO TRANSCRIPTION AND ELECTROPORATION OF MRNA DNA encoding the respective transgene was amplified by PCR and
subsequently transcribed in vitro with the mMessage mMachine T7 Ultra Kit (Ambion) according to the manufacturer’s instructions. The resulting mRNA was purified using the RNeasy Kit
(Qiagen). Electroporation was performed with <1 × 107 cells/100 µl Opti-MEM (without phenol red; Thermo Scientific) using a Square wave protocol (1 pulse, 500 V) on the Gene Pulser Xcell
Electroporation System (Biorad) and 4 mm electroporation cuvettes (VWR). The length of the pulse was 5 ms for primary human T cells and 3 ms for Jurkat cells. Primary human T cells were
electroporated with 5 µg of CAR-mRNA and Jurkat cells were electroporated with different mRNA amounts as indicated. After electroporation, the cells were immediately recovered in warm cell
growth medium and incubated over night at 37 °C. LENTIVIRAL TRANSDUCTION To generate VSV-G pseudotyped lentivirus, Lenti-X 293T cells (Takara) were co-transfected with a third-generation
puromycin-selectable pCDH expression vector (System Biosciences) and second-generation viral packaging plasmids pMD2.G and psPAX2 (Addgene plasmids #12259 and #12260, respectively; gifts
from Didier Trono) using the PureFection Transfection Reagent (System Biosciences) according to the manufacturer’s instructions. Supernatants were collected on day 2 and 3 after transfection
and were concentrated 100-fold using the Lenti-X Concentrator (Takara) according to the instructions provided by the manufacturer. Viral suspensions were resuspended in RPMI-1640 medium
supplemented with 10% FCS and 1% penicillin–streptomycin and frozen at −80 °C. Twenty-four hours after their activation with αCD3/αCD28 beads (Thermo Scientific), primary human T cells were
transduced in cell culture plates, which were coated according to the manufacturer’s instructions with RetroNectin (Takara). Thawed viral supernatant was added to the T cells (0.5 × 106
cells/mL) at a final dilution of 1:2. To ensure high and uniform expression of the transgenes, 1 µg/mL puromycin (Sigma-Aldrich) was added 3 days later and removed after 2 more days.
Transduced T cells were expanded in AIMV medium (Thermo Scientific) supplemented with 2% Octaplas (Octapharma), 1% l-Glutamine (Thermo Scientific), 2.5% HEPES (Thermo Scientific), and 200
U/mL recombinant human IL-2. Half of the medium was exchanged every 2–3 days. Cell lines were transduced by exposure to varying concentrations of lentiviral supernatants for 3 days, followed
by puromycin selection for 2 days with varying concentrations of puromycin and expansion in the respective growth medium. FLOW CYTOMETRIC ANALYSIS Cells were counted using AccuCheck
Counting Beads (Thermo Scientific). Propidium iodide (Sigma-Aldrich) was added for exclusion of dead cells. Unspecific antibody binding was prevented by preincubation of the cells for 10 min
at 4 °C in FACS buffer [i.e., PBS (Thermo Scientific), 0.2% human albumin (CSL Behring) and 0.02% sodium azide (Merck KGaA)] supplemented with 10% (v/v) human serum. Alternatively, for
subsequent detection of Fc-containing CARs, preincubation was done in antihuman FACS buffer (i.e., PBS, 10% FCS and 0.02% sodium azide) supplemented with 10% (v/v) murine IgG1-κ (clone
MOPC21; 1 mg/mL; Sigma-Aldrich). This preincubation was omitted for the detection of the FMC63-derived αCD19-CAR by the protein L-biotin conjugate (GenScript; dilution 1:167). The following
antibodies were used: αFLAG tag (PE; clone L5; BioLegend; dilution 1:167), αFLAG tag (APC; clone L5; BioLegend; dilution 1:167), αStrep II tag (biotin; clone 5A9F9; GenScript; dilution
1:500), αhEGFR (APC and PE; clone AY13; BioLegend; dilution 1:50), and αhHER2 (PE; clone 24D2; BioLegend; dilution 1:50), αhCD19 (BV421; clone HIB19, BioLegend; dilution 1:167), αhCD45
(PerCP; clone 2D1; BD Biosciences; dilution 1:50), αhCD3 (PE-Cy7; clone SK7; BD Biosciences; dilution 1:50), αGFP (PE; clone FM264G; BioLegend; dilution 1:50). Binding of hEGFR-Fc and
mEGFR-Fc (both R&D) to hEGFR-specific CARs was detected by an antibody directed against human IgG1-Fc (PE; clone JDC-10; Southern Biotech; dilution 1:25). Cells were incubated with the
respective antibodies or protein L for 25 min at 4 °C and then washed twice with ice-cold FACS buffer or—if Fc-containing CARs were detected—antihuman FACS buffer. Biotinylated antibodies
and protein L were further incubated with streptavidin APC or streptavidin PE (both Thermo Scientific; dilution 1:250) for 25 min at 4 °C and then again washed twice. For the analysis of
single-cell suspensions of mouse organs, dead cells were excluded by staining with the Fixable Viability Dye eFluor™ 780 (Thermo Scientific). Subsequently, the cells were acquired on an LSR
Fortessa instrument (BD Biosciences) and analyzed using the FlowJo Software (Version 10.6.1; FlowJo, LLC) and the BD FACSDivaTM Software V8.0.1. Nontransfected cells or isotype antibodies
served as negative controls. QUANTITATION OF TARGET ANTIGEN DENSITY The surface density of target antigen molecules on Jurkat cells was quantified using the QuantiBRITE Phycoerythrin (PE)
Fluorescence Quantitation Kit (Becton Dickinson) according to the manufacturer´s instructions. Briefly, the cells were stained with a saturating concentration of a PE-labeled αhEGFR-antibody
(clone AY13; BioLegend). Subsequently, the geometric mean of the fluorescence intensity was determined, subjected to background subtraction using unstained or isotype-labeled cells and then
used to estimate the number of antibodies bound per cell (ABC). ABC values were corrected for the PE-conjugation efficiency of the respective antibody (i.e., PE dye-to-antibody ratio)
yielding effective surface densities. GENERATION OF MUTANT BINDERS BY ALANINE SCANNING Site-directed mutagenesis of the hEGFR- and hHER2-specific binder scaffolds was performed using the
QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Genomics), according to the manufacturer’s instructions. Mutagenic primers were designed using the QuikChange Primer Design
Software (Agilent Genomics) and synthesized by Biomers, Germany. Primer sequences can be found in Supplementary Data 3. In the case of the hHER2-specific binder, the affibody zHER2-AK was
generated from the parental affibody zHER2:4, which binds to hHER2 with an affinity of 50 nM45, prior to the alanine scan by introducing mutations N23A and S33K for preventing
N-glycosylation66. DETERMINATION OF BINDING AFFINITIES ON CELL MEMBRANES Jurkat cells with high level expression of hEGFRt were obtained by electroporation with 3 µg of hEGFRt-mRNA. On the
next day, the cells were washed with PBS, resuspended in PBSA [PBS supplemented with 0.1% BSA (Sigma-Aldrich)] and incubated in 96-well plates for 1 h at 4 °C with varying concentrations of
fluorescent chimeric hEGFR-binder proteins (i.e., E11.4.1 or mutants thereof fused to sfGFP). The plates were then centrifuged (450 × _g_, 7 min, 4 °C), the supernatant discarded and the
cells resuspended in ice-cold PBSA immediately prior to acquisition on an LSR Fortessa instrument. The cells were kept on ice during the analysis to avoid endocytosis. _K_d values were
calculated by curve fitting using Microsoft Excel (Microsoft Corporation). CONSTRUCTION OF TRANSGENES A detailed description of all used transgenes is given in Supplementary Data 1. hEGFRt
and hHER2t were generated using the Addgene plasmids #1101167 and #1625768, respectively (gifts from Matthew Meyerson and Mien-Chie Hung). sfGFP was derived from the Addgene plasmid #5473769
(gift from Michael Davidson & Geoffrey Waldo). Cytoplasmic domains of CD28, ICOS, OX40, and CD2 were derived from cDNA clones (Sino Biological). The cytoplasmic domain of CD3ζ contained
a Q65K mutation (Uniprot P20963-3). Synthetic DNAs were synthesized by GeneArt (Thermo Scientific). Assembly of DNA molecules into complete constructs was performed using the Gibson
Assembly Master Mix (New England BioLabs) according to the manufacturer’s instructions. RECOMBINANT EXPRESSION OF BINDER PROTEINS To determine their affinities, the binders were expressed
recombinantly as soluble proteins with an N-terminal hexahistidine and a C-terminal sfGFP fusion tag using the pE-SUMO vector (Life Sensors), in which the SUMO-tag was deleted. Briefly,
_Escherichia coli_ cells (Tuner DE3) were transformed with respective plasmids using heat shock transformation. After overnight incubation in lysogeny broth (LB) medium at 37 °C, cultures
were diluted 1:100 in terrific broth medium supplemented with kanamycin (50 µg/mL) and were kept shaking at 37 °C. Expression of the transgene was started when cultures reached an A600 of 2
by addition of 1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) followed by overnight incubation at 20 °C. Transformed cells were harvested by centrifugation (5,000 × _g_, 20 min, 4 °C)
and resuspended in sonication buffer (50 mM sodium phosphate, 300 mM NaCl, 3% glycerol, 1% Triton X-100, pH 8.0). Cell disruption was accomplished by sonication (2 × 90 s, duty cycle 50%,
amplitude set to 5) followed by removal of cell debris by centrifugation (20,000 × _g_, 30 min, 4 °C). Hexahistidine-tagged proteins were extracted from crude cell lysates by immobilized
metal affinity chromatography using TALON metal affinity resin (Takara). Sonicated supernatants were supplemented with 10 mM imidazole and applied onto the resin twice, followed by repeated
washing steps with equilibration buffer (50 mM sodium phosphate, 300 mM NaCl, pH 8.0) supplemented with increasing concentrations of imidazole (5–15 mM). Purified proteins were eluted by
washing the column with equilibration buffer with 250 mM imidazole. Buffer exchange to PBS was performed with Amicon Ultra-15 10 K centrifugal filters (Merck Millipore). Affibody-based
binder scaffolds were further purified by performing a preparative SEC using a Superdex 200 column (10 mm × 300 mm, GE Healthcare). Purified proteins were directly frozen at −80 °C.
Truncated human VEGF (Uniprot P15692 amino acids 40–134) was expressed as previously described46. Briefly, _E. coli_ BL21 (DE3) cells were transformed with the pJ414 vector (ATUM) including
the gene encoding for truncated VEGF. LB medium supplemented with ampicillin (100 µg/mL) was inoculated with an overnight culture. Once the culture reached an A600 of 2 at 37 °C, the
overexpression was induced by addition of 1 mM IPTG. After 4 h of shaking at 37 °C, cells were harvested by centrifugation (5,000 × _g_, 20 min, 4 °C). The pellet was resuspended in 20 mM
TRIS-HCl buffer (pH 7.5) including 5 mM ethylenediaminetetraacetic acid (EDTA), incubated for 10 min and disrupted by ultrasonication with a Vibra-Cell 375 ultrasonic processor (Sonics &
Materials, Inc.). After two washing steps using the same buffer, the pellet was resuspended in 20 mL unfolding buffer (20 mM TRIS-HCl, 5 mM EDTA, 7.5 M urea, 4 mM dithiothreitol, pH 7.5),
stirred for 2 h at room temperature followed by centrifugation (39,000 × _g_, 25 min, 4 °C). The supernatant containing the unfolded protein was diluted tenfold into refolding buffer (20 mM
TRIS-HCl, 7 mM CuCl2, pH 8.4) and stirred overnight at room temperature. Following a dialysis step against 20 mM TRIS-HCl (pH 8.0), the protein was purified using a 6 mL Resource Q column, a
1 mL HiTrap Phenyl FF (Low Sub) column and a HiLoad 16/600 Superdex 200 pg (all from GE Healthcare) according to the manufacturer’s instructions. Pure VEGF samples were stored at −80 °C.
SIZE EXCLUSION CHROMATOGRAPHY For analytical SEC analysis, recombinant proteins were diluted in SEC running buffer (PBS supplemented with 200 mM NaCl) and filtered through a 0.1 µm Ultrafree
MC VV centrifugal filter (Merck Millipore). Subsequently, 25 µg protein was loaded onto a Superdex 200 column (10 mm × 300 mm, GE Healthcare) connected to an HPLC Prominence LC20 System
(Shimadzu) at a flow rate of 0.75 mL/min at 25 °C. SURFACE PLASMON RESONANCE SPR analysis was performed with a Biacore T200 instrument (GE Healthcare). All experiments were performed in
degassed and filtered PBS, pH 7.4, supplemented with 0.1% BSA (Sigma-Aldrich) and 0.05% Tween-20 (Merck Millipore) at 25 °C. Immobilization of hEGFR-Fc and hHER2-Fc (both R&D) was
performed on a Protein A sensor chip (GE Healthcare) at a flow rate of 10 µL/min for 60 s at a concentration of 6.67 and 4 µg/mL, respectively, yielding a density of ~400 response units.
Five different concentrations of the respective binder scaffold (depending on the expected _K_d value) were injected at a flow rate of 30 µL/min for 15 s (for binders E11.4.1, E11.4.1-G25A,
E11.4.1-G32A, and zHER2-AK-R10A) or 60 s (for zHER2-AK) in the single-cycle kinetic mode. Subsequently, a dissociation step was performed for 30 s (all E11.4.1-based binders), 60 s
(zHER2-AK-R10A), or 180 s (zHER2-AK). Protein A sensor chips were regenerated by applying 10 mM Glycine-HCl, pH 1.5, at a flow rate of 30 µL/min for 30 s. The _K_d value was either obtained
by steady state binding analysis or by fitting the sensorgram to a 1:1 Langmuir model in the kinetic mode using the Biacore T200 Evaluation Software (GE Healthcare). WESTERN BLOTTING AND
CROSSLINKING OF MEMBRANE PROTEINS Crosslinking of membrane proteins was performed with disuccinimidyl suberate (DSS) (Thermo Scientific) according to the manufacturer’s instructions.
Briefly, Jurkat cells were washed three times with ice-cold PBS. Cells were resuspended in PBS to a cell concentration of 20 × 106 cells/mL. Subsequently, DSS was added at a concentration of
5 mM and the reaction was incubated for 30 min at room temperature. Chemical crosslinking was stopped by addition of the Quench Solution (1 M TRIS, pH 7.5), yielding a final concentration
of 20 mM TRIS. Cells were washed once in PBS and lysed in ice-cold RIPA buffer (Sigma-Aldrich), supplemented with protease inhibitors (Halt Protease Inhibitor Cocktail; Thermo Scientific)
and nucleases (Pierce Universal Nuclease; Thermo Scientific). The lysate was incubated on ice for 5 min. Subsequently, samples were denatured for 5 min at 50 °C in loading buffer (LDS Sample
Buffer; Thermo Scientific) and frozen at −20 °C. Cleared lysates (14,000 × _g_, 10 min, 4 °C) were resolved on 4–20% gradient SDS-PAGE gels (Mini-PROTEAN TGX Precast Protein Gels; BioRad)
under nonreducing conditions. Separated proteins were transferred to 0.45 µm nitrocellulose membrane (GE Healthcare) and blocking was performed with Western Blocking Reagent (Roche) for 1 h
at room temperature. Membranes were probed with an antibody against hEGFR (clone 528; 1:500 dilution; Thermo Scientific) and incubated overnight at 4 °C. Subsequently, membranes were
incubated with a secondary goat antimouse horseradish peroxidase-conjugated antibody (Thermo Scientific) for 1 h at room temperature and bands were developed using the SuperSignal West Femto
Maximum Sensitivity Substrate (Thermo Scientific), according to the manufacturer’s instructions. Loading control was performed by probing the membrane with an antibody against GAPDH (clone
ab9485; 1:5,000; Abcam) for 1 h at room temperature. Detection was performed by incubating the membrane with a secondary goat antirabbit DyLight 800-conjugated antibody (Thermo Scientific)
for 1 h at room temperature and visualizing the bands on the Odyssey Imaging System with the Odyssey Infrared Imaging System software (Version 3.0, LI-COR). CYTOTOXICITY ASSAY Cytotoxicity
of primary human T cells was assayed with either a luciferase- or a flow cytometry-based assay. Luciferase-based assay: 10,000 target cells (i.e., luciferase-expressing Nalm-6 or Jurkat
cells) were co-cultured for 4 h at 37 °C with 20,000 primary human T cells (= E:T cell ratio 2:1, unless stated otherwise) in white round-bottom 96-well plates (Sigma-Aldrich) in 100 µL RPMI
medium without phenol red (Thermo Scientific) supplemented with 10% FCS and 1% penicillin–streptomycin. After co-culture, viable target cells were quantified by determining the luciferase
activity. Hereto, the culture plates were equilibrated for 10 min to room temperature and subsequently luciferin (Xenolight D-luciferin; Perkin Elmer) was added to the cell suspension (150
µg/mL final concentration). Bioluminescence was quantified after 20 min of incubation at room temperature on an ENSPIRE Multimode plate reader (Perkin Elmer). Specific lysis was calculated
with the following formula: $$\% \,{\mathrm{specific}}\,{\mathrm{lysis}} = 100 - \left( {\frac{{\frac{{{\mathrm{RLU}}\,{\mathrm{antigen}}^{{\mathrm{pos}}}{\mathrm{target}} \, + \,
{\mathrm{effector}}\,{\mathrm{cells}}}}{{{\mathrm{RLU}}\,{\mathrm{antigen}}^{{\mathrm{pos}}}{\mathrm{target}}\,{\mathrm{cells}}\,{\mathrm{only}}}}}}{{\left(
{\frac{{{\mathrm{RLU}}\,{\mathrm{antigen}}^{{\mathrm{neg}}}{\mathrm{target}} \, + \,
{\mathrm{effector}}\,{\mathrm{cells}}}}{{{\mathrm{RLU}}\,{\mathrm{antigen}}^{{\mathrm{neg}}}{\mathrm{target}}\,{\mathrm{cells}}\,{\mathrm{only}}}}} \right)}}} \right) \times 100.$$ (1)
Specific lysis in Fig. 5a, b was calculated with the following formula: $${\mathrm{\% }}\,{\mathrm{specific}}\,{\mathrm{lysis}} = 100 - \left( {\frac{{{\mathrm{RLU}}\,{\mathrm{target}} +
{\mathrm{effector}}\,{\mathrm{cells}}}}{{\left( {{\mathrm{RLU}}\,{\mathrm{target}}\,{\mathrm{cells}}\,{\mathrm{only}}} \right)}}} \right) \times 100.$$ (2) Flow cytometry-based cytotoxicity
assay: the assay principle is based on the determination of the ratio of antigenpos and antigenneg target cells that expressed either a green or red fluorescent protein, respectively, and
were mixed 1:1 before co-culture with T cells. For this purpose, Jurkat cells were electroporated on the day before the assay (1) with mRNA encoding eGFP and mRNA encoding the respective
target antigen, and alternatively (2) with mRNA encoding mCherry. Those cells were co-cultured for 4 h at 37 °C in round-bottom 96-well plates with 40,000 primary human T cells at an E:T
cell ratio of 4:1:1 (i.e., 40,000 CAR-T cells plus 10,000 antigenpos and 10,000 antigenneg Jurkat cells per well). Target cells not co-cultured with CAR-T cells served as a negative control
(targets only). After co-culture, the ratio of eGFP- to mCherry-expressing target cells was determined by flow cytometric analysis. Specific lysis was calculated with the following formula:
$$\% \,{\mathrm{specific}}\,{\mathrm{lysis}} = 100 - \left( {\frac{{\frac{{\% \,{\mathrm{eGFP}}^{{\mathrm{pos}}}\,{\mathrm{cells}}\,{\mathrm{of}}\,{\mathrm{the}}\,{\mathrm{sample}}}}{{\%
\,{\mathrm{mCherry}}^{{\mathrm{pos}}}\,{\mathrm{cells}}\,{\mathrm{of}}\,{\mathrm{the}}\,{\mathrm{sample}}}}}}{{\left( {\frac{{\%
\,{\mathrm{eGFP}}^{{\mathrm{pos}}}\,{\mathrm{cells}}\,{\mathrm{of}}\, {\hbox{''}}{\mathrm{targets}}\,{\mathrm{only}}{\hbox{''}} \,{\mathrm{control}}}}{{\%
\,{\mathrm{mCherry}}^{{\mathrm{pos}}}\,{\mathrm{cells}}\,{\mathrm{of}}\, {\hbox{''}}{\mathrm{targets}}\,{\mathrm{only}}{\hbox{''}} \,{\mathrm{control}}}}} \right)}}}
\right) \times 100.$$ (3) CYTOKINE SECRETION BY CAR-T CELLS Cytokine secretion of CAR-T cells was determined by co-cultivation with target cells at an E:T cell ratio of 2:1 in flat-bottom
96-well plates for 4 h at 37 °C. Alternatively, cytokine secretion was quantified in the supernatants from cytotoxicity experiments, described above. Supernatants were centrifuged (450 ×
_g_, 7 min, 4 °C) and stored at −80 °C. IFN-γ was quantified by enzyme-linked immunosorbent assay (ELISA) using the Human IFN-γ ELISA Ready-SET-Go! Kit (eBioscience/Thermo Scientific)
according to the manufacturer’s instructions. Analysis was performed on the ENSPIRE Multimode plate reader (Perkin Elmer). IN VIVO EXPERIMENTS NOD.Cg-Prkdcscid Il2rgtm1WJI/SzJ (NSG, The
Jackson Laboratory) mice were bred in the Anna Spiegel facility for animal breeding (Vienna, Austria) under standardized conditions (room temperature 22 ± 2 °C, humidity 55 ± 10%, air change
rate of 15 and a dark/light cycle of 12 h). Subsequently, mice were transferred to the Preclinical Imaging Laboratory (PIL) or the Center for Biomedical Research of the Medical University
of Vienna. All procedures were approved by the Magistratsabteilung 58, Vienna, Austria (GZ: 319093/2014/16) and the Federal Ministry Republic of Austria for Education, Science, and Research
(BMBWF-66.009/0243-V/3b/2019). Primary human T cells were lentivirally transduced for CAR expression and expanded for 15–17 days to generate sufficient cell numbers. CAR-T cells directed
against hEGFR showed no cross-reactivity with mEGFR as confirmed with recombinant mEGFR-Fc (R&D) (Supplementary Fig. 7a). Nalm-6 cells were engineered to express high levels of the
hEGFRt, hHER2t, or both (intracellularly fused to the wild-type FKBP12 domain) and ffLuc [hEGFRt-Nalm-6 (SEQ-ID 20), hHER2t-Nalm-6 (SEQ-ID 23), and hHER2t-hEGFRt-Nalm-6 (SEQ-ID 20 and 23),
respectively]. After filtering through a 35 µm cell strainer (Corning Falcon), 0.5 × 106 of those Nalm-6 cells (in 100 µL PBS) were injected i.v. into the tail veins of NSG mice (male and
female, 8–20 weeks of age). Three or 5 days later (as indicated), when tumor burden became detectable by bioluminescence imaging (BLI), CAR-T cells (10 × 106 cells in 100 µL PBS) were
injected i.v. into the tail veins of the NSG mice. Dimerization of the ON-switch AvidCAR was induced by intraperitoneal (i.p.) administration of 2 mg/kg AP20187 (Takara). AP20187 was
prepared by dissolution in ethanol yielding a concentration of 12.5 mg/mL and further dilution to the final working concentration of 0.5 mg/mL in vehicle solution [4% ethanol, 10% PEG-400
and 1.7% Tween-80 (both Sigma-Aldrich) in water]. Those working stocks of AP20187 were sterile filtered and used for injection within 30 min. Injection of only the vehicle solution served as
control. The mice were monitored daily and sacrificed by cervical dislocation at the first sign of disease (weight loss, rough fur, beginning paralysis). IN VIVO BIOLUMINESCENCE IMAGING BLI
was performed at the PIL (Medical University of Vienna) using an IVIS Spectrum In Vivo Imaging System (Perkin Elmer). D-luciferin (Perkin Elmer) was freshly dissolved in PBS to a final
concentration of 15 mg/mL and sterile filtered. Mice were anesthetized with isoflurane (Abbvie) and received i.p. injections of the luciferin solution (dosage of 150 mg/kg body weight).
After 15–20 min, mice were transferred to the IVIS Imaging System and bioluminescence was measured immediately in medium binning mode with an automatic acquisition time (ranging from 1 s to
2 min) to obtain unsaturated images. The total photon flux was determined within the region of interest which encompassed the entire body of the mouse. Tumor growth was monitored every 3–4
days. Living Image Software (Caliper) was used to analyse the data. IN VIVO MODEL FOR THE AND-GATE AVIDCAR NSG mice were injected via tail vein with 0.5 × 106 cells of a 1:1:1:1 mixture of
Nalm-6, hEGFRt-Nalm-6, hHER2t-Nalm-6 and hHER2t-hEGFRt-Nalm-6 cells and 3 days later with 10 × 106 CAR-T cells or mock-T cells, as indicated. Mice were sacrificed 13 days after tumor
injection. Bone marrow single-cell suspensions were obtained by flushing one femur with ~10 mL sterile PBS and passing through a 70 µm cell strainer (Corning Falcon). After disruption of the
liver through a 70 µm cell strainer and several washing steps, lymphocytes were separated from hepatocytes using a 33.75% Percoll gradient (GE Healthcare). Spleen single-cell suspensions
were obtained by two consecutive passages through 70 µm cell strainers. Blood was drawn retro-orbitally from isoflurane-anesthesized mice and collected in EDTA-containing tubes (Greiner).
Fifty µL blood was used for the staining with fluorescent antibodies, followed by red blood cell lysis using an RBC lysis/fixation solution (Biolegend). MATHEMATICAL MODELING The multivalent
interaction between a bivalent antigen and a bivalent CAR that can undergo kinetic proofreading leads to a large number of distinct chemical states. Therefore, it is not practical to
manually enumerate them. To overcome this, we used the rule-based framework of BioNetGen70 to define five elementary reaction: intermolecular binding (_k_on), intermolecular unbinding
(_k_off), intramolecular binding (_σ k_on), and the kinetic proofreading phosphorylation rate (_k_p). We have assumed that (1) the intramolecular unbinding rate (i.e., the unbinding rate
between a single CAR and antigen when both CARs are bound to antigen) is equal to the intermolecular unbinding (i.e., unbinding rate of a single CAR and antigen when only a single CAR in the
dimer is bound to antigen) and (2) the kinetic proofreading phosphorylation rate can only take place when at least one CAR in the dimer is bound to antigen. When a single CAR in the dimer
is bound, the binding rate of the second CAR in the dimer to the second antigen in the dimer is expected to be dependent on the intermolecular on-rate (since the same binding interface is
formed) multiplied by a factor that accounts for the local concentration (and potentially other factors, such as structural alignment) that we define to be σ with units of concentration.
This parameter controls the avidity of the CAR with _σ_ = 0 producing a monovalent CAR and increasing values of _σ_ lead to increasing avidity. Collectively, these elementary reactions lead
BioNetGen to 72 chemical states that were exported as a system of 72 coupled nonlinear coupled ordinary differential equations. The initial conditions were 0 for all chemical species except
the concentration of the CAR (1,000/μm2) and antigen (varied as indicated). The parameter values used were _k_on = 5 × 10−4 μm/s, _k_off = 10/s, and _k_p = 0.1/s. These equations were solved
numerically in Matlab (Mathworks, MA) to produce the panels in Supplementary Fig. 3b showing the bound receptor and phosphorylated receptor. To calculate the cellular response in
Supplementary Fig. 3b, we have assumed a saturating Hill function connecting phosphorylated receptor to the cellular response using a threshold of (_k_thres = 2/μm2). Although the parameter
values are expected to alter the absolute concentration of antigen inducing receptor binding, phosphorylation, and cellular response, they do not alter our main qualitative conclusion which
is that increasing the cooperativity factor of dimeric binding (_σ_) increases the amount of phosphorylated receptor and the cellular response without increasing CAR binding. The bngl file
defining the model along with the m-file used to solve it can be found in Supplementary Data 4 that also includes all parameter values. STATISTICAL ANALYSIS Statistical analysis was
performed using GraphPad Prism 7 software for Windows (GraphPad Software Inc.) and SAS software (Version 9.4, SAS Institute). Graphs were generated using GraphPad Prism 7 software. Data are
presented as individual data points or as means ± SD. Statistical analysis was performed using two-tailed paired _t-_tests, two-tailed ratio-paired _t_-test, or two-tailed unpaired _t_-test,
as indicated. Statistical analysis of Figs. 4b and 6b was done with a mixed model to compare fixed-effect means taking into account that the data are correlated. Statistical analysis of
Figs. 5d and 6d was done by log-rank analysis or two-way ANOVA, respectively. REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary
linked to this article. DATA AVAILABILITY The authors declare that all data supporting the findings of this study are available within the article and its Supplementary information files or
from the corresponding authors upon reasonable request. The source data for the Figs. 2a, c, e, 3b, e, g, 4b, d, 5a–c, and 6b, d, as well as Supplementary Figs. 2c, 4a, b, d, 5e, 6a, b, and
12c, e are provided as a Source data file. Source data are provided with this paper. REFERENCES * Labanieh, L., Majzner, R. G. & Mackall, C. L. Programming CAR-T cells to kill cancer.
_Nat. Biomed. Eng._ 2, 377–391 (2018). CAS PubMed Google Scholar * June, C. H. & Sadelain, M. Chimeric antigen receptor therapy. _N. Engl. J. Med._ 379, 64–73 (2018). CAS PubMed
Google Scholar * Morgan, R. A. et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. _Mol.
Ther._ 18, 843–851 (2010). CAS PubMed PubMed Central Google Scholar * Parkhurst, M. R. et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal
cancer but induce severe transient colitis. _Mol. Ther._ 19, 620–626 (2011). CAS PubMed Google Scholar * Lamers, C. H. J. et al. Treatment of metastatic renal cell carcinoma with CAIX
CAR-engineered T cells: clinical evaluation and management of on-target toxicity. _Mol. Ther._ 21, 904–912 (2013). CAS PubMed PubMed Central Google Scholar * Linette, G. P. et al.
Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. _Blood_ 122, 863–871 (2013). CAS PubMed PubMed Central Google Scholar * Morgan,
R. A. et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. _J. Immunother._ 36, 133–151 (2013). CAS PubMed PubMed Central Google Scholar * Roybal,
Kole T. et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. _Cell_ 164, 770–779 (2016). CAS PubMed PubMed Central Google Scholar * Fedorov, V. D.,
Themeli, M. & Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. _Sci. Transl. Med._ 5, 215ra172 (2013). PubMed
PubMed Central Google Scholar * Cho, J. H., Collins, J. J. & Wong, W. W. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. _Cell_ 173,
1426–1438.e1411 (2018). CAS PubMed PubMed Central Google Scholar * Sukumaran, S. et al. Enhancing the potency and specificity of engineered T cells for cancer treatment. _Cancer Discov._
8, 972–987 (2018). CAS PubMed PubMed Central Google Scholar * Wilkie, S. et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide
complementary signaling. _J. Clin. Immunol._ 32, 1059–1070 (2012). CAS PubMed Google Scholar * Kloss, C. C., Condomines, M., Cartellieri, M., Bachmann, M. & Sadelain, M. Combinatorial
antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. _Nat. Biotechnol._ 31, 71–75 (2012). PubMed PubMed Central Google Scholar *
Lanitis, E. et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. _Cancer Immunol. Res._
1, 43–53 (2013). CAS PubMed PubMed Central Google Scholar * Liu, X. et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against
tumors in mice. _Cancer Res._ 75, 3596–3607 (2015). CAS PubMed PubMed Central Google Scholar * Caruso, H. G. et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal
tissue while maintaining potent antitumor activity. _Cancer Res._ 75, 3505–3518 (2015). CAS PubMed PubMed Central Google Scholar * Arcangeli, S. et al. Balance of anti-CD123 chimeric
antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. _Mol. Ther._ 25, 1933–1945 (2017). CAS PubMed PubMed Central Google Scholar * Majzner, R. G. et
al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. _Clin. Cancer Res._ 25, 2560–2574 (2019). *
Han, C. et al. Desensitized chimeric antigen receptor T cells selectively recognize target cells with enhanced antigen expression. _Nat. Commun._ 9, 468 (2018). ADS PubMed PubMed Central
Google Scholar * Chmielewski, M., Hombach, A., Heuser, C., Adams, G. P. & Abken, H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment
domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. _J. Immunol._ 173, 7647–7653 (2004). CAS PubMed Google Scholar
* Wu, C.-Y., Roybal, K. T., Puchner, E. M., Onuffer, J. & Lim, W. A. Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. _Science_ 350, aab4077
(2015). ADS PubMed PubMed Central Google Scholar * Leung, W.-H. et al. Sensitive and adaptable pharmacological control of CAR T cells through extracellular receptor dimerization. _JCI
Insight_, https://doi.org/10.1172/jci.insight.124430 (2019). * Juillerat, A. et al. Design of chimeric antigen receptors with integrated controllable transient functions. _Sci. Rep._ 6,
18950 (2016). ADS CAS PubMed PubMed Central Google Scholar * Hill, Z. B., Martinko, A. J., Nguyen, D. P. & Wells, J. A. Human antibody-based chemically induced dimerizers for cell
therapeutic applications. _Nat. Chem. Biol._ 14, 112–117 (2018). CAS PubMed Google Scholar * Urbanska, K. et al. A universal strategy for adoptive immunotherapy of cancer through use of a
novel T-cell antigen receptor. _Cancer Res._ 72, 1844–1852 (2012). CAS PubMed PubMed Central Google Scholar * Ma, J. S. Y. et al. Versatile strategy for controlling the specificity and
activity of engineered T cells. _Proc. Natl. Acad. Sci. USA_ 113, E450–E458 (2016). CAS PubMed Google Scholar * Rodgers, D. T. et al. Switch-mediated activation and retargeting of CAR-T
cells for B-cell malignancies. _Proc. Natl. Acad. Sci. USA_ 113, E459–E468 (2016). CAS PubMed Google Scholar * Cartellieri, M. et al. Switching CAR T cells on and off: a novel modular
platform for retargeting of T cells to AML blasts. _Blood Cancer J._ 6, e458 (2016). CAS PubMed PubMed Central Google Scholar * Zajc, C. U. et al. A conformation-specific ON-switch for
controlling CAR T cells with an orally available drug. _Proc. Natl. Acad. Sci. USA_ 117, 14926–14935 (2020). CAS PubMed Google Scholar * Srivastava, S. et al. Logic-gated ROR1 chimeric
antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. _Cancer Cell_ 35, 489–503.e488 (2019). CAS PubMed PubMed Central
Google Scholar * Aruffo, A. & Seed, B. Molecular cloning of a CD28 cDNA by a high-efficiency COS cell expression system. _Proc. Natl. Acad. Sci. USA_ 84, 8573–8577 (1987). ADS CAS
PubMed Google Scholar * Hennecke, S. & Cosson, P. Role of transmembrane domains in assembly and intracellular transport of the CD8 molecule. _J. Biol. Chem._ 268, 26607–26612 (1993).
CAS PubMed Google Scholar * Liu, H. & May, K. Disulfide bond structures of IgG molecules. _MAbs_ 4, 17–23 (2012). CAS PubMed PubMed Central Google Scholar * Long, A. H. et al.
4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. _Nat. Med._ 21, 581–590 (2015). CAS PubMed PubMed Central Google Scholar *
Atwell, J. L. et al. scFv multimers of the anti-neuraminidase antibody NC10: length of the linker between VH and VL domains dictates precisely the transition between diabodies and
triabodies. _Protein Eng. Des. Sel._ 12, 597–604 (1999). CAS Google Scholar * Whitlow, M., Filpula, D., Rollence, M. L., Feng, S.-L. & Wood, J. F. Multivalent Fvs: characterization of
single-chain Fv oligomers and preparation of a bispecific Fv. _Protein Eng. Des. Sel._ 7, 1017–1026 (1994). CAS Google Scholar * Nieba, L., Honegger, A., Krebber, C. & Plückthun, A.
Disrupting the hydrophobic patches at the antibody variable/constant domain interface: improved in vivo folding and physical characterization of an engineered scFv fragment. _Protein Eng.
Des. Sel._ 10, 435–444 (1997). CAS Google Scholar * Traxlmayr, M. W. et al. Strong enrichment of aromatic residues in binding sites from a charge-neutralized hyperthermostable Sso7d
scaffold library. _J. Biol. Chem._ 291, 22496–22508 (2016). CAS PubMed PubMed Central Google Scholar * Tischer, D. K. & Weiner, O. D. Light-based tuning of ligand half-life supports
kinetic proofreading model of T cell signaling. _eLife_ 8, e42498 (2019). PubMed PubMed Central Google Scholar * James, J. R. Tuning ITAM multiplicity on T cell receptors can control
potency and selectivity to ligand density. _Sci. Signal._ 11, eaan1088 (2018). PubMed PubMed Central Google Scholar * Ajina, A. & Maher, J. Strategies to address chimeric antigen
receptor tonic signaling. _Mol. Cancer Ther._ 17, 1795–1815 (2018). CAS PubMed PubMed Central Google Scholar * Grada, Z. et al. TanCAR: a novel bispecific chimeric antigen receptor for
cancer immunotherapy. _Mol. Ther.—Nucl. Acids_ 2, e105 (2013). Google Scholar * Hegde, M. et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. _J. Clin.
Invest._ 126, 3036–3052 (2016). PubMed PubMed Central Google Scholar * De Munter, S. et al. Nanobody based dual specific CARs. _Int. J. Mol. Sci._ 19, 403 (2018). * Wikman, M. et al.
Selection and characterization of HER2/neu-binding affibody ligands. _Protein Eng. Des. Sel._ 17, 455–462 (2004). CAS PubMed Google Scholar * Lobner, E. et al. Two-faced Fcab prevents
polymerization with VEGF and reveals thermodynamics and the 2.15 Å crystal structure of the complex. _MAbs_ 9, 1088–1104 (2017). CAS PubMed PubMed Central Google Scholar * Moll, J. R.,
Ruvinov, S. B., Pastan, I. & Vinson, C. Designed heterodimerizing leucine zippers with a ranger of pIs and stabilities up to 10−15 M. _Protein Sci._ 10, 649–655 (2001). CAS PubMed
PubMed Central Google Scholar * Stone, J. D., Harris, D. T. & Kranz, D. M. TCR affinity for p/MHC formed by tumor antigens that are self-proteins: impact on efficacy and toxicity.
_Curr. Opin. Immunol._ 33, 16–22 (2015). CAS PubMed PubMed Central Google Scholar * Fridy, P. C. et al. A robust pipeline for rapid production of versatile nanobody repertoires. _Nat.
Methods_ 11, 1253–1260 (2014). CAS PubMed PubMed Central Google Scholar * Shinozaki, N., Hashimoto, R., Fukui, K. & Uchiyama, S. Efficient generation of single domain antibodies with
high affinities and enhanced thermal stabilities. _Sci. Rep._ 7, 5794 (2017). ADS PubMed PubMed Central Google Scholar * Arndt, K. M., Müller, K. M. & Plückthun, A. Factors
influencing the dimer to monomer transition of an antibody single-chain Fv fragment. _Biochemistry_ 37, 12918–12926 (1998). CAS PubMed Google Scholar * Wörn, A. & Plückthun, A.
Stability engineering of antibody single-chain Fv fragments. _J. Mol. Biol._ 305, 989–1010 (2001). PubMed Google Scholar * McKeithan, T. W. Kinetic proofreading in T-cell receptor signal
transduction. _Proc. Natl. Acad. Sci. USA_ 92, 5042–5046 (1995). ADS CAS PubMed Google Scholar * Yousefi, O. S. et al. Optogenetic control shows that kinetic proofreading regulates the
activity of the T cell receptor. _eLife_ 8, e42475 (2019). PubMed PubMed Central Google Scholar * Goldstein, B., Faeder, J. R. & Hlavacek, W. S. Mathematical and computational models
of immune-receptor signalling. _Nat. Rev. Immunol._ 4, 445–456 (2004). CAS PubMed Google Scholar * Dobri, N. et al. A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic
bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. _Investig. Ophthalmol. Vis. Sci._ 54, 85–95 (2013). CAS Google Scholar * Kovacs, E., Zorn, J. A., Huang, Y.,
Barros, T. & Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. _Annu. Rev. Biochem._ 84, 739–764 (2015). CAS PubMed PubMed Central Google
Scholar * Gasparrini, F. et al. Nanoscale organization and dynamics of the siglec CD22 cooperate with the cytoskeleton in restraining BCR signalling. _EMBO J._ 35, 258–280 (2016). CAS
PubMed Google Scholar * Polyak, M. J., Li, H., Shariat, N. & Deans, J. P. CD20 homo-oligomers physically associate with the B Cell antigen receptor: dissociation upon receptor
engagement and recruitment of phosphoproteins and calmodulin-binding proteins. _J. Biol. Chem._ 283, 18545–18552 (2008). CAS PubMed Google Scholar * Kennedy, S. P., Hastings, J. F., Han,
J. Z. R. & Croucher, D. R. The under-appreciated promiscuity of the epidermal growth factor receptor family. _Front. Cell Dev. Biol._ 4, 88 (2016). PubMed PubMed Central Google Scholar
* Kufe, D. W. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. _Oncogene_ 32, 1073–1081 (2013). CAS PubMed Google Scholar *
Cazet, A. et al. The ganglioside G(D2) induces the constitutive activation of c-Met in MDA-MB-231 breast cancer cells expressing the G(D3) synthase. _Glycobiology_ 22, 806–816 (2012). CAS
PubMed Google Scholar * Liang, Y.-J. et al. Interaction of glycosphingolipids GD3 and GD2 with growth factor receptors maintains breast cancer stem cell phenotype. _Oncotarget_ 8,
47454–47473 (2017). * Jacobson, K., Liu, P. & Lagerholm, B. C. The lateral organization and mobility of plasma membrane components. _Cell_ 177, 806–819 (2019). CAS PubMed PubMed
Central Google Scholar * Garcia-Parajo, M. F., Cambi, A., Torreno-Pina, J. A., Thompson, N. & Jacobson, K. Nanoclustering as a dominant feature of plasma membrane organization. _J.
Cell Sci._ 127, 4995–5005 (2014). PubMed PubMed Central Google Scholar * Feldwisch, J. et al. Design of an optimized scaffold for affibody molecules. _J. Mol. Biol._ 398, 232–247 (2010).
CAS PubMed Google Scholar * Greulich, H. et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. _PLoS Med._ 2, e313 (2005). PubMed PubMed Central Google
Scholar * Li, Y. M. et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. _Cancer Cell_ 6, 459–469 (2004). CAS PubMed Google Scholar * Pédelacq, J.-D., Cabantous,
S., Tran, T., Terwilliger, T. C. & Waldo, G. S. Engineering and characterization of a superfolder green fluorescent protein. _Nat. Biotechnol._ 24, 79–88 (2006). PubMed Google Scholar
* Harris, L. A. et al. BioNetGen 2.2: advances in rule-based modeling. _Bioinformatics_ 32, 3366–3368 (2016). CAS PubMed PubMed Central Google Scholar Download references
ACKNOWLEDGEMENTS We would like to acknowledge Ulrike Pötschger for assistance in statistical analysis. The SPR equipment was kindly provided by the EQ-BOKU VIBT GmbH and the BOKU Core
Facility for Biomolecular & Cellular Analysis. We are thankful to the excellent support of the Preclinical Imaging Laboratory (PIL) and the Center for Biomedical Research of the Medical
University of Vienna. This work is supported by the Austrian Science Fund (FWF Project P32001-B and FWF Project W1224—Doctoral Program on Biomolecular Technology of Proteins—BioToP), the
Federal Ministry for Digital and Economic Affairs of Austria, and the National Foundation for Research, Technology and Development of Austria to the Christian Doppler Research Association
(Christian Doppler Laboratory for Next Generation CAR T Cells) and by private donations to the Children’s Cancer Research Institute (Vienna, Austria). O.D. is supported by a Wellcome Trust
Senior Research Fellowship (207537/Z/17/Z). T.P. and J.B.H. were supported by the European Unionʼs Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant
agreement no. 721358. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * St. Anna Children’s Cancer Research Institute (CCRI), 1090, Vienna, Austria Benjamin Salzer, Christina M. Schueller,
Charlotte U. Zajc, Michael A. Schoeber, Boris Kovacic, Michelle C. Buri, Eva M. Putz, Wolfgang Holter & Manfred Lehner * Christian Doppler Laboratory for Next Generation CAR T Cells,
1090, Vienna, Austria Benjamin Salzer, Charlotte U. Zajc, Michael W. Traxlmayr & Manfred Lehner * Center for Pathophysiology, Infectiology and Immunology, Institute for Hygiene and
Applied Immunology, Medical University of Vienna, 1090, Vienna, Austria Timo Peters & Johannes B. Huppa * Department of Biotechnology, University of Natural Resources and Life Sciences,
1190, Vienna, Austria Elisabeth Lobner * Sir William Dunn School of Pathology, University of Oxford, Oxford, OX1 3RE, UK Omer Dushek * Department of Chemistry, Institute of Biochemistry,
University of Natural Resources and Life Sciences, 1190, Vienna, Austria Christian Obinger & Michael W. Traxlmayr * Department of Pediatrics, St. Anna Kinderspital, Medical University of
Vienna, 1090, Vienna, Austria Wolfgang Holter & Manfred Lehner Authors * Benjamin Salzer View author publications You can also search for this author inPubMed Google Scholar * Christina
M. Schueller View author publications You can also search for this author inPubMed Google Scholar * Charlotte U. Zajc View author publications You can also search for this author inPubMed
Google Scholar * Timo Peters View author publications You can also search for this author inPubMed Google Scholar * Michael A. Schoeber View author publications You can also search for this
author inPubMed Google Scholar * Boris Kovacic View author publications You can also search for this author inPubMed Google Scholar * Michelle C. Buri View author publications You can also
search for this author inPubMed Google Scholar * Elisabeth Lobner View author publications You can also search for this author inPubMed Google Scholar * Omer Dushek View author publications
You can also search for this author inPubMed Google Scholar * Johannes B. Huppa View author publications You can also search for this author inPubMed Google Scholar * Christian Obinger View
author publications You can also search for this author inPubMed Google Scholar * Eva M. Putz View author publications You can also search for this author inPubMed Google Scholar * Wolfgang
Holter View author publications You can also search for this author inPubMed Google Scholar * Michael W. Traxlmayr View author publications You can also search for this author inPubMed
Google Scholar * Manfred Lehner View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS B.S., C.M.S., C.U.Z., T.P., and M.A.S. performed the
experiments and analyzed data. B.S., E.M.P., J.B.H., M.W.T, and M.L. designed the experiments and critically revised the data. B.K., M.C.B., and E.M.P. assisted in animal models. E.L.
contributed valuable reagents and helped with experimental design. O.D. assisted in mathematical modeling. B.S., O.D., C.O., W.H., M.W.T., and M.L. wrote the paper. All authors edited and
approved the paper. CORRESPONDING AUTHORS Correspondence to Michael W. Traxlmayr or Manfred Lehner. ETHICS DECLARATIONS COMPETING INTERESTS B.S., C.U.Z., C.O., M.W.T., and M.L. have filed
three patent applications that relate to the technology described in this paper: patent applicant: St. Anna Kinderkrebsforschung (Zimmermannplatz 10, 1090 Vienna, Austria), Universität für
Bodenkultur Wien (Gregor Mendel-Strasse 33, 1180 Vienna, Austria); name of inventor(s): B.S., M.W.T., M.L.; application number: PCT/EP2019/076917 (WO 2020/070290 A1); status of application:
pending; specific aspect of paper covered in patent application: AvidCARs with inducible function. Patent applicant: St. Anna Kinderkrebsforschung (Zimmermannplatz 10, 1090 Vienna, Austria),
Universität für Bodenkultur Wien (Gregor Mendel-Strasse 33, 1180 Vienna, Austria); name of inventor(s): B.S., M.W.T., M.L;. application number: PCT/EP2019/076916 (WO 2020/070289 A1); status
of application: pending; specific aspect of paper covered in patent application: AvidCARs for combinatorial antigen recognition (AND-gate function). Patent applicant: St. Anna
Kinderkrebsforschung (Zimmermannplatz 10, 1090 Vienna, Austria), Universität für Bodenkultur Wien (Gregor Mendel-Strasse 33, 1180 Vienna, Austria); name of inventor(s): Charlotte Brey*,
M.T., C.O., M.L.; application number: PCT/EP2018/086299 (WO 2019/122188 A1); status of application: pending; specific aspect of paper covered in patent application: alternative ON-switch
system for conditional heterodimerization of AvidCARs (mentioned in the “Discussion”) (*now C.U.Z.). The remaining authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW
INFORMATION _Nature Communications_ thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. PUBLISHER’S NOTE Springer
Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION SUPPLEMENTARY DATA 1
SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 SUPPLEMENTARY DATA 4 PEER REVIEW FILE REPORTING SUMMARY DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS
OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or
other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Salzer, B., Schueller,
C.M., Zajc, C.U. _et al._ Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. _Nat Commun_ 11, 4166 (2020).
https://doi.org/10.1038/s41467-020-17970-3 Download citation * Received: 19 August 2019 * Accepted: 23 July 2020 * Published: 20 August 2020 * DOI: https://doi.org/10.1038/s41467-020-17970-3
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy
to clipboard Provided by the Springer Nature SharedIt content-sharing initiative