Heterozygous rare variants in nr2f2 cause a recognizable multiple congenital anomaly syndrome with developmental delays
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Nuclear receptor subfamily 2 group F member 2 (_NR2F2_ or _COUP-TF2_) encodes a transcription factor which is expressed at high levels during mammalian development. Rare
heterozygous Mendelian variants in _NR2F2_ were initially identified in individuals with congenital heart disease (CHD), then subsequently in cohorts of congenital diaphragmatic hernia (CDH)
and 46,XX ovotesticular disorders/differences of sexual development (DSD); however, the phenotypic spectrum associated with pathogenic variants in _NR2F2_ remains poorly characterized.
Currently, less than 40 individuals with heterozygous pathogenic variants in _NR2F2_ have been reported. Here, we review the clinical and molecular details of 17 previously unreported
individuals with rare heterozygous _NR2F2_ variants, the majority of which were de novo. Clinical features were variable, including intrauterine growth restriction (IUGR), CHD, CDH, genital
anomalies, DSD, developmental delays, hypotonia, feeding difficulties, failure to thrive, congenital and acquired microcephaly, dysmorphic facial features, renal failure, hearing loss,
strabismus, asplenia, and vascular malformations, thus expanding the phenotypic spectrum associated with _NR2F2_ variants. The variants seen were predicted loss of function, including a
nonsense variant inherited from a mildly affected mosaic mother, missense and a large deletion including the _NR2F2_ gene. Our study presents evidence for rare, heterozygous _NR2F2_ variants
causing a highly variable syndrome of congenital anomalies, commonly associated with heart defects, developmental delays/intellectual disability, dysmorphic features, feeding difficulties,
hypotonia, and genital anomalies. Based on the new and previous cases, we provide clinical recommendations for evaluating individuals diagnosed with an _NR2F2_-associated disorder. You have
full access to this article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS EXPANDING THE PHENOTYPIC SPECTRUM AND CLINICAL SEVERITY ASSOCIATED WITH _WLS_ GENE
Article 28 April 2023 BIALLELIC _PAN2_ VARIANTS IN INDIVIDUALS WITH A SYNDROMIC NEURODEVELOPMENTAL DISORDER AND MULTIPLE CONGENITAL ANOMALIES Article 18 March 2022 HAPLOINSUFFICIENCY OF
_SF3B2_ CAUSES CRANIOFACIAL MICROSOMIA Article Open access 03 August 2021 INTRODUCTION Nuclear receptor subfamily 2 group F member 2 (_NR2F2_, also known as _COUP-TF2_) gene encodes a member
of the nuclear receptor superfamily of ligand-activated transcriptional factors involved in several developmental and cellular processes. NR2F2 is an orphan receptor whose ligand is yet to
be identified. Similar to the other nuclear receptors, NR2F2 protein has three main domains: an N-terminal activation binding motif (1-78aa), a DNA-binding domain (79-151aa), and a
C-terminal ligand-binding domain (177-411aa) separated by the hinge region [1, 2]. The spatiotemporal expression pattern of _NR2F2_ during mammalian development has been studied in mouse
models [1, 3, 4]. _Nr2f2_ expression is observed between E8.5 and E13.5 in the sinus venosus, umbilical veins, heart, atrium, branchial arches, developing hindbrain, neuroectoderm of
anterior midbrain, somites, otocyst, the periocular mesenchyme, optic stalk, olfactory placode, developing testes, the genital tubercle, mesenchyme of the kidney, and the adrenal cortex [1,
4]. High expression of _NR2F2_ is observed in the mesenchymal component of several organs during development and organogenesis [1, 3]. Homozygous deletion of _Nr2f2_ is embryonic lethal in
mice. The _Nr2f2_−/− embryos show growth restriction, edematous cysts, severe hemorrhage in the brain and heart, and die around 10 days of gestation [4]. Heterozygous _Nr2f2_ knockout mice
are smaller than the wild-type mice and show poor postnatal viability. Furthermore, _Nr2f2_ knockout mice show defects in sinus venosus development, dysplastic anterior and posterior
cardinal veins, atrial malformations, and angiogenesis defects [4]. A recent study showed that female mouse embryos lacking _Nr2f2_ in the Wolffian duct mesenchyme develop as intersex, that
is, having both female and male reproductive tracts [5]. Other _Nr2f2_ mouse models showed developmental anomalies of the female reproductive system [6,7,8], anteroposterior patterning of
the stomach [9], diaphragm [10], lymphangiogenesis, adipogenesis, limb, eye, and cerebellum [3]. In humans, _NR2F2_ is extremely intolerant to loss‐of‐function (LoF) variants (pLI = 0.99 in
Genome Aggregation Database (gnomAD) v2.1.1) [11, 12]. _NR2F2_ heterozygous LoF variants have been associated with congenital malformations, including CHD, CDH, and DSD (Supplementary Table
1) [1, 13,14,15,16,17,18,19,20,21,22,23,24]. However, most of these studies were done using disease-specific cohorts and thus have limited clinical description, especially about the other
affected organ systems. Al Turki et al. (2014) described eight individuals with CHD who had rare LoF, missense, and splice variants, and a balanced translocation in _NR2F2_ (Supplementary
Table 1) [1]. Subsequently, _NR2F2_ rare variants were reported in other CHD cohorts [14,15,16,17, 22]. High et al. (2016) identified two individuals with CDH and heterozygous LoF variants
in _NR2F2_ [24], other CDH-cohort studies have also identified rare _NR2F2_ LoF variants and a splice variant [15, 19, 21, 24, 25]. In a cohort of 46,XX SRY-negative ovotesticular DSD cases,
Bashamboo et al. (2018) found _NR2F2_ LoF variants in three individuals with syndromic clinical features including CHD, CDH and dysmorphic facial features of blepharophimosis, ptosis, and
epicanthus inversus syndrome (BPES) [20]. Recently, a rare _NR2F2_ de novo missense variant was reported in an individual with 46,XY DSD, micropenis and hypospadias [26]. Two case reports
have described small chromosome 15q26.2 deletions (<5Mb) encompassing _NR2F2_: a de novo mosaic 1.7Mb deletion in a fetus with CDH and coarctation of the aorta, and a de novo 3Mb
heterozygous deletion in a 46,XX individual reared as male with ovotesticular DSD, dysmorphic features, sinus bradycardia, low weight, blepharophimosis, and ptosis (Supplementary Table 1)
[27, 28]. Larger deletions (>5Mb) involving _NR2F2_ along with other genes [29,30,31,32,33,34] and a 15q26.2 deletion including _NR2F2_ but without precise molecular breakpoints have also
been reported [35]. Here, we describe 17 previously unreported individuals with varied phenotypes, including CHD, CDH, and other affected organ systems including vascular malformations, who
have rare heterozygous, mostly de novo variants in _NR2F2_. We review and compare the clinical and molecular data of these new cases with previously reported individuals, to delineate and
expand the phenotypic and genotypic spectrum of _NR2F2_-associated disorders. MATERIALS AND METHODS Trio whole exome sequencing (WES) was performed for the cases, except for case 8 as the
father’s testing was pending and case 12, where chromosomal microarray analysis was performed. Detailed methods are provided in the Supplementary information. RESULTS MOLECULAR FINDINGS
Fifteen of the seventeen newly reported individuals in our cohort had de novo, rare, heterozygous variants in _NR2F2_ (NM_021005.4; NP_066285.1), and one (individual 5-1) had a variant
inherited from a mosaic, mildly affected mother (individual 5-2, 34% variant allelic fraction, Supplementary Table 2 and Fig. 1). The inheritance for the remaining two individuals was
unknown. Sixteen of the seventeen individuals had a heterozygous _NR2F2_ single nucleotide variant (SNV) whereas one case (individual 12) had a large deletion. Ten of the fifteen unique
_NR2F2_ SNVs were rare missense variants; the remaining five were predicted LoF variants. All _NR2F2_ variants identified in this study were absent in gnomAD (v2.1.1 and v3) and TOPMed
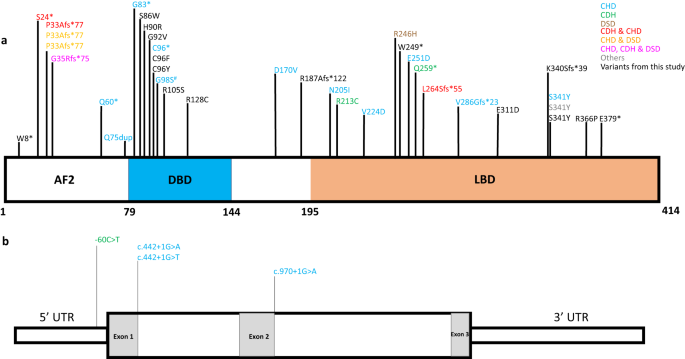
population databases (see Table 1 for details on all variants). The distribution of _NR2F2_ variants from our study and previously reported cases on the encoded protein and the gene are
shown in Fig. 1 and Supplementary Fig. 1. Notably, two predicted LoF variants (p.Lys340SerfsTer39, individual 4; p.Glu379Ter, individual 13) map to the last coding exon of _NR2F2_ and may
escape nonsense-mediated decay; no other downstream truncating variants are reported in the affected individuals. These truncations delete a part of the ligand binding domain; however,
whether they lead to the loss of NR2F2 function is currently unknown. We also reviewed whole exome sequencing data from the Pediatric Cardiac Genomics Consortium (PCGC) cohort [36, 37],
which consists of individuals with cardiac defects and identified one additional case with a rare de novo _NR2F2_ variant (p.Gly98Ser). This case is not included in the current cohort, but
has been shown in Fig. 1. In one individual (individual 12) chromosomal microarray identified a novel, large de novo deletion (~1.87Mb, arr[GRCh37] 15q26.2q26.3(96755103_98628389)x1 at
chromosome 15q26.2 encompassing _NR2F2-AS1_, _NR2F2, SPATA8-AS1, LINC02254, LINC00923, ARRDC4_, and _LINC01582_ genes (Supplementary Fig. 1). This deletion results in the loss of the entire
_NR2F2_ gene and this loss is not seen in the population databases (Database of Genomic Variants (DGV), gnomAD SVs v2.1). _ARRDC4_, the only other protein-coding gene in this deleted region,
is not constrained for LoF variants (pLI = 0, gnomAD v2) and currently has no known disease associations. Therefore, the clinical features observed in this individual were ascribed to the
single copy loss of _NR2F2_. CLINICAL FINDINGS We describe 16 unrelated affected individuals and a mildly affected mosaic mother of one of the individuals with heterozygous variants in
_NR2F2_. Clinical case reports for the 17 individuals are described in the Supplementary Information. These individuals had variable clinical features, including developmental
delays/intellectual disability, CHDs, dysmorphic facial features, feeding difficulties, hearing impairment, hypotonia, genital anomalies, renal abnormalities, CDH, vascular malformations and
prenatal findings, including CHD, IUGR, increased nuchal translucency, and single umbilical artery. The clinical features of these individuals are described in Supplementary Table 2 and the
frequencies of the phenotypes observed in our cohort are listed in Table 2. The commonly observed clinical phenotypes in multiple individuals with _NR2F2_ variants in our cohort are
described below. PRENATAL FINDINGS CHDs were detected prenatally in four of seventeen (23.5%) individuals whereas eight of the remaining individuals (47%) had prenatal findings other than
CHD, including IUGR, increased nuchal translucency, single umbilical artery, oligohydramnios, hepatic vascular malformation and CDH (Supplementary Table 2). POSTNATAL FINDINGS FEEDING
DIFFICULTIES Fourteen of seventeen (82%) individuals had feeding difficulties and/ most with a history of nasogastric tube dependence (13/14). DEVELOPMENTAL DELAYS/INTELLECTUAL DISABILITY
All 14 individuals for whom data were available had motor delays and thirteen of them also had speech delays. Delays were noted for the age at sitting, walking, and utterance of first words.
Hypotonia was observed in 59% (10/17) of the cohort. Two of the thirteen individuals (15%) for whom brain imaging data were available, showed periventricular white matter abnormalities. CHD
All individuals (17/17) in our cohort had CHD. The commonly seen anomalies included atrial septal defect (8/17), coarctation of the aorta (8/17), ventricular septal defect (5/17), aortic
arch anomalies (3/17), atrioventricular septal defect (2/17), dilated right ventricle (2/17) and pulmonary hypertension (2/17). CDH CDH was observed in two of seventeen (12%) individuals,
and both died within a few days after birth. DYSMORPHIC FEATURES Characteristic facial features were observed in all individuals except for the mosaic mother of individual 5 (16/17; 94%).
The commonly observed anomalies (seen in ≥3 affected individuals) included upslanted or short palpebral fissures, hypertelorism, low-set and/or dysplastic ears, full cheeks, and retrognathia
or micrognathia (Supplementary Tables 2, 4 and Supplementary Fig. 2). Other facial features which were seen in any 2 affected individuals were microcephaly, bilateral epicanthus, deeply set
eyes, long eyelashes, long philtrum, facial asymmetry, dysplastic ears, and short neck. Some of the less common features included synophrys, highly arched eyebrows, ptosis, down turned
corners of mouth, and tapered finger among others (Supplementary Tables 2, 4). HEARING LOSS Five of fifteen (33%) individuals had hearing loss, which was unilateral in four cases. At least
one individual needed hearing aids and another individual had conductive hearing loss. STRABISMUS Divergent strabismus was seen in 6 of 15 (40%) individuals. GENITAL ANOMALIES & DSD Six
of seven 46,XY males (86%) in the cohort had cryptorchidism. Out of the nine 46,XX females, one had labial hypertrophy. One of the 46,XX individuals was diagnosed with DSD (individual 15,
SRY negative) and had a de novo _NR2F2_ LoF variant (c.23G>A, p.(Trp8Ter)). Prenatally, aberrant right subclavian artery, IUGR, and oligohydramnios were detected in this individual and
amniocentesis revealed a 46,XX karyotype. At birth, male external genitalia with perineal hypospadias, pigmented bifid scrotum with penoscrotal transposition and inguinal gonads were
observed. There was no history of maternal virilization during the pregnancy. This 46,XX karyotype individual was diagnosed with DSD, severe aortic coarctation, and aberrant right subclavian
artery. The abdominal ultrasound showed the presence of both testes, and a testis biopsy revealed testicular parenchyma comprising seminiferous ducts made up of Sertoli cells without the
presence of spermatogonia, and the presence of Leydig cells in the testicular interstitium. Subsequently, bilateral orchidopexy, urethroplasty, penoscrotal transposition correction, and
scrotal closure were performed. RENAL ANOMALIES Six of seventeen (35%) individuals had renal findings including renal failure (2/17), renal hypoplasia (1/17), and multicystic dysplastic
kidneys (1/17). One of the individuals who had renal hypoplasia and congenital renal failure underwent a renal transplant. Nephrocalcinosis was also noted in one person. ASPLENIA Asplenia
was noted in two of seventeen (12%) individuals. VASCULAR MALFORMATIONS Vascular malformations were seen in two of seventeen (12%) individuals, including one with a hepatic vascular
malformation and another with vascular lesions on skin. SKELETAL ABNORMALITIES Five out of seventeen (29%) individuals had 5th finger clinodactyly; talipes and clawing of toes were each
noted in two individuals, and other skeletal findings were seen in seven out of seventeen individuals (41%; Supplementary Table 2). DISCUSSION In this study, we describe 17 previously
unreported individuals with rare, potentially disease causing variants in _NR2F2_ and provide a comprehensive delineation of the clinical and molecular features of the associated syndrome
(Tables 1 and 2, Fig. 1, Supplementary Figs. 3 and 4 and Supplementary Tables 2 and 3). The clinical and molecular details of all the previously reported cases with rare heterozygous _NR2F2_
variants are summarized in Supplementary Table 1. MOLECULAR SPECTRUM Fourteen of the fifteen _NR2F2_ SNVs detected in our cohort are novel variants and have not been previously reported.
Nine individuals described in our cohort carry novel rare missense _NR2F2_ variants. The missense variant seen in the Pediatric Cardiac Genomics Consortium case (p.(Gly98Ser)) is also not
reported before [36, 37]. An additional six missense variants have been previously reported in affected individuals, one of which is recurrent in our cohort (p.(Ser341Tyr); Fig. 1 and
Supplementary Table 1). These variants localize to the two conserved functional domains of NR2F2 protein, the DNA-binding domain and the ligand-binding domain (Fig. 1 and Supplementary Figs.
3 and 4). Al Turki et al. [1] used an in vitro luciferase assay in HEK293 cells to show the functional effects of _NR2F2_ missense variants found in the CHD cohorts (p.(Asp170Val),
p.(Asn205Ile), p.(Glu251Asp), p.(Ser341Tyr), and p.(Ala412Ser)). Luciferase assays using transfected expression plasmids of the _NR2F2_ variants showed a significant change in the
transcriptional activity when compared with wild type _NR2F2_ plasmid; the direction of this effect (increased or decreased transcriptional activity) was dependent on the promoter context,
that is, _NGFI-A_ or _APOB_ promoters which are known direct targets for _NR2F2_ binding [1]. These results along with the presence of _NR2F2_ LoF variants in the affected individuals
suggest that the global mis-regulation of transcriptional activities of _NR2F2_ targets due to haploinsufficiency or LoF or possibly gain-of-function variants in _NR2F2_ during development,
may lead to congenital anomalies in multiple organ systems. Most of the reported _NR2F2_ missense variants, however, have not been functionally characterized. Notably, _NR2F2_ is constrained
for missense changes (missense Z score = 3.6, gnomADv2.1.1). The pathogenicity of the _NR2F2_ missense variants detected in the affected individuals is supported by their absence in the
population databases, the de novo status, and the pathogenic in silico predictions of their functional impact. In the future, assessing the effect of the _NR2F2_ missense variants using in
vitro luciferase assays for the transcriptional activity or in vivo chromatin immune-precipitation (ChIP) experiments using patient-derived cells to assess the changes in levels of NR2F2
protein binding to target genomic regions, such as gene promoters or regulatory regions, will further elucidate the mechanism of these missense alterations. CLINICAL FEATURES The most common
clinical features observed in our cohort included developmental delays/intellectual disability (100%), CHD (100%), dysmorphic features (94.1%), feeding difficulties or feeding tube
dependence (82.4%), hypotonia (58.8%) and cryptorchidism in 46,XY males (86%, Table 2 and Supplementary Table 3). All individuals had developmental delays, specifically, language was delayed
in all individuals, with a wide range for first words (11 months to ‘not yet achieved’ at 8 years). Motor milestones were also delayed, with walking achieved at 2–3 years in some cases, but
‘not yet achieved’ at 8 years in others. All the affected individuals reported here had CHD and extensive additional medical and developmental issues. The types of heart defects observed
were similar to those reported previously, with the majority of individuals having either an atrial or ventricular septal defect or coarctation of the aorta. Three individuals had aortic
arch anomalies and two individuals had atrioventricular septal defects. Other less common cardiac defects were valvular and supravalvular pulmonic stenosis, aberrant left subclavian artery,
tricuspid regurgitation and bicuspid aortic valve. Two individuals had pulmonary hypertension and four had cardiac arrhythmias. Interestingly, a de novo _NR2F2_ variant p.(Ser341Tyr) was
recently reported in a male with developmental delays, asplenia, frequent respiratory infections, dysmorphic facial features, bilateral cryptorchidism, and glandular hypospadias but with no
associated CHD, CDH or DSD (Supplementary Table 1) [13]. Echocardiograms and abdominal ultrasound ruled out the presence of CHD and CDH in this individual. Nearly all individuals in our
cohort had dysmorphic facial features, most common being upslanted or short palpebral fissures, micrognathia or retrognathia, low-set or dysplastic ears, hypertelorism, and full cheeks.
Other features seen were microcephaly, bilateral epicanthus, long eyelashes, long philtrum, facial asymmetry and short neck. Dysmorphic facial features seen in our cohort and in previously
reported individuals with _NR2F2_ pathogenic variants [16, 18, 20]; [Supplementary Table 1] overlap with the findings seen in the BPES which is caused by heterozygous loss of function
variants in _FOXL2_ gene. The shared BPES phenotype in affected individuals with _FOXL2_/_NR2F2_ pathogenic variants suggests, these genes might function together in the developmental
pathways leading to eyelid formation. Similarly, both these transcription factors are known to function in mammalian ovarian development and are potential “pro-ovary/anti-testis” genes [20,
38]. Other common findings include feeding difficulties, which required requiring gastric tube due to growth issues. Clinical features that have not been commonly associated with _NR2F2_
variants in previously published cases, were seen in our cohort. Five individuals had hypoplastic or dysplastic kidneys, including two with subsequent chronic renal failure, five had hearing
impairment, six had strabismus, and three individuals had sleep apnea or breath holding. Laryngomalacia and accessory spleen were also seen in two individuals each (Supplementary Table 2).
Two related individuals (5-1 and 5-2) had asplenia or polysplenia, a phenotypic feature that was recently reported in another affected individual with a de novo _NR2F2_ variant [13].
Interestingly, foregut mesenchyme specific conditional _Nr2f2_−/− mice were previously shown to have increased perinatal mortality, CDH, and spleen defects including asplenia [10]. Also,
vascular malformations, a phenotype seen in the _NR2F2_ mouse model [4], was seen in two individuals of our cohort (individuals 4 and 13). _Nr2f2_−/− mice showed defects in angiogenesis and
vascular remodeling, including enlarged blood vessels, abnormal development of the atria and sinus venosus, malformed cardinal veins, and decrease in the complexity of microvasculature in
the head and spine regions [4]. Differential diagnoses that were considered for these individuals included Noonan syndrome, Prader-Willi syndrome, Pitt-Hopkins syndrome, Fryns syndrome,
Smith-Lemli-Opitz syndrome, peroxisomal disorders, Russell-Silver syndrome, geleophysic dysplasia, Moore Federman syndrome, Myhre syndrome, DSD, and laterality defects. GENOTYPE-PHENOTYPE
CORRELATION Review of all known _NR2F2_ SNVs, did not demonstrate any genotype-phenotype correlation or hotspot region(s) where missense variants were significantly enriched (Fig. 1).
However, many missense variants are localized to a narrow region in the DNA binding domain (aa 86–105; Fig. 1). Interestingly, the 46,XX DSD associated _NR2F2_ LoF variants were all early
truncations in coding exon 1 (individual 15, [20]; Fig. 1]) or entire gene deletion [27], whereas, on the contrary, all the affected 46,XX individuals with _NR2F2_ early truncations [16, 23]
or gene deletion (individual 12) did not present with a DSD phenotype (Fig. 1 and Supplementary Table 1). Our cohort had five predicted LoF _NR2F2_ SNVs, and they were present in
individuals with a syndromic CHD presentation with neurodevelopmental issues (individuals 5-1, 6, 10 and 13), or 46,XX ovotesticular DSD (individual 15). Similar to our cohort, _NR2F2_ LoF
variants have been previously reported in cases with CHD, CDH, and syndromic 46,XX ovotesticular DSD (Supplementary Table 1) [1, 13,14,15,16,17,18,19,20,21,22,23,24,25]. In our cohort, there
were ten 46,XX individuals, nine of whom were phenotypic females whereas one individual, who carried a heterozygous _NR2F2_ LoF variant, was assigned male gender at birth and had a
diagnosis of ovotesticular DSD (individual 15; Table 1). _NR2F2_ LoF variants and a _NR2F2_ gene deletion have been previously reported in individuals with 46,XX ovotesticular DSD [20, 27].
However, individual 12, a 46,XX phenotypic female, who carried a single copy genomic deletion encompassing the entire _NR2F2_ gene, did not show evidence of genital anomalies or DSD, but
presented with CHD, developmental delays, feeding difficulties, and dysmorphic features. A similar case with a _NR2F2_ gene deletion has been reported [28]. The three known deletion CNVs
encompassing _NR2F2_ also had varied phenotypes, including syndromic CHD (individual 12), syndromic DSD without CHD [27], and syndromic CHD with CDH [28] (Supplementary Fig. 1). These
clinical phenotypes highlight the variable expressivity in _NR2F2_-associated disorders. In previous studies the CDH phenotype has only been associated with de novo _NR2F2_ LoF SNVs [15, 21,
23, 24] or gene deletion [27], whereas in our cohort, de novo _NR2F2_ missense variants in the DNA binding domain were seen in the two individuals affected with CDH (individuals 10 and 11).
A comparison of phenotypes between our cases with missense variants in the DNA binding domain (individuals 2,3,7,8,10,11,14) and in the ligand binding domain (individuals 1,9,16) did not
yield any genotype phenotype correlation. Larger cohorts may be needed to recognize any association. Two of the _NR2F2_ variants in our cohort affect amino acid Cys96 (p.(Cys96Phe),
p.(Cys96Tyr)), and a nonsense variant at the same amino acid residue has been reported in a CHD cohort [17]. p.(Cys96Ter) variant was identified in a family with six affected individuals
with bicuspid aortic valve and fusion of the right and left coronary aortic valve cusps. Two of the affected individuals had aortic stenosis along with aortic regurgitation and another two
affected individuals had VSD in addition to bicuspid aortic valve. The two affected individuals in our cohort (individuals 2 and 8) with de novo variants altering Cys98, had CHD,
developmental delays, feeding difficulties, and dysmorphic facial features. Individual 2 also had multicystic dysplastic kidneys with chronic renal failure. Interestingly, two _NR2F2_
variants have each been observed recurrently in three unrelated, affected individuals (p.(Pro33AlafsTer77) and p.(Ser341Tyr)). The p.(Pro33AlafsTer77) LoF variant has been reported in two
individuals with CHD and 46,XX ovotesticular DSD and in one individual with CHD and CDH [20, 23]. For one of the individuals with 46,XX ovotesticular DSD, the inheritance of
p.(Pro33AlafsTer77) was unknown, and for the remaining two cases it was de novo. Similarly, the de novo p.(Ser341Tyr) variant was seen in one individual with an atrioventricular septal
defect [1] and in a second individual with asplenia, developmental delays, dysmorphic features, frequent infections, bilateral cryptorchidism, and glandular hypospadias without CHD [13]. In
our cohort, this variant was seen in an individual with an atrioventricular defect, feeding difficulties, developmental delays and dysmorphic facial features (individual 9); however, the
inheritance of this variant was unknown (not maternal). These variable phenotypic manifestations in individuals carrying identical variants underscore the variable expressivity associated
with _NR2F2_ sequence alterations. Additional genetic and environmental modifiers may have played a role in the phenotypic expression of the _NR2F2_-related disorder. Based on the clinical
features described in our study, we recommend that any individual with a pathogenic/likely pathogenic _NR2F2_ variant should have the following evaluations (1) echocardiogram and
electrocardiogram (2) renal-bladder ultrasound (3) assessment of external genitalia and consideration of imaging if ambiguous (pelvic ultrasound for females/genital tract ultrasound for
males) (4) audiology evaluation (5) ophthalmology examination (6) consideration of early placement of gastric tube for problems with weight gain and oral feeding (7) early intervention for
speech, occupational, and physical therapy (8) neuropsychological evaluation and developmental assessment as soon as diagnosis is made and (9) confirmation of sex chromosome complement.
Testing for _NR2F2_ should be considered in any syndromic CHD patient, especially with BPES facial features. In conclusion, we report findings of 17 unreported individuals and summarize
previously reported individuals with heterozygous variants in _NR2F2_, to provide a comprehensive overview of the spectrum of associated features. Based on these data, we provide clinical
recommendations for assessment of affected individuals upon diagnosis. These data and recommendations provide guidance for the clinicians and laboratory personnel who encounter this rare
disorder. DATA AVAILABILITY The data in this study is available in the manuscript, Supplementary information, tables, and figures. The _NR2F2_ variants reported in this study are submitted
in ClinVar. The following variants have been previously submitted by the clinical testing labs/research groups in ClinVar NM_021005.4: c.1019del:p.(Lys340SerfsTer39),
c.746G>A:p.(Trp249Ter), c.558dup:p.(Arg187AlafsTer122), c.269A>G:p.(His90Arg), c.1022C>A:p.(Ser341Tyr), c.1097G>C:p.(Arg366Pro) (Variation IDs: 2429770, 598763, 1805610, 1064859,
128232, 521133). The details of the two variants - c.287G>T:p.(Cys96Phe), c.257C>G:p.(Ser86Trp) which were ascertained as part of the DDD study are available in the DECIPHER website
(https://www.deciphergenomics.org/patient/259383/genotype/191257/browser; https://www.deciphergenomics.org/patient/282004/genotype/197133/browser). The Clinvar IDs for the remaining variants
are listed below (Variation IDs: 2570648, 2570646, 2570643, 2570644, 2570645, 2570642, 2570647). REFERENCES * Al Turki S, Manickaraj AK, Mercer CL, Gerety SS, Hitz MP, Lindsay S, et al.
Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet. 2014;94:574–85. Article CAS PubMed PubMed Central Google Scholar * Polvani S, Pepe S, Milani S, Galli A.
COUP-TFII in health and disease. Cells. 2019;9:101. Article PubMed PubMed Central Google Scholar * Lin FJ, Qin J, Tang K, Tsai SY, Tsai MJ. Coup d'Etat: an orphan takes control.
Endocr Rev. 2011;32:404–21. Article CAS PubMed PubMed Central Google Scholar * Pereira FA, Qiu Y, Zhou G, Tsai MJ, Tsai SY. The orphan nuclear receptor COUP-TFII is required for
angiogenesis and heart development. Genes Dev. 1999;13:1037–49. Article CAS PubMed PubMed Central Google Scholar * Zhao F, Franco HL, Rodriguez KF, Brown PR, Tsai MJ, Tsai SY, et al.
Elimination of the male reproductive tract in the female embryo is promoted by COUP-TFII in mice. Science. 2017;357:717–20. Article CAS PubMed PubMed Central Google Scholar * Kurihara
I, Lee DK, Petit FG, Jeong J, Lee K, Lydon JP, et al. COUP-TFII mediates progesterone regulation of uterine implantation by controlling ER activity. PLoS Genet. 2007;3:e102. Article PubMed
PubMed Central Google Scholar * Petit FG, Jamin SP, Kurihara I, Behringer RR, DeMayo FJ, Tsai MJ, et al. Deletion of the orphan nuclear receptor COUP-TFII in uterus leads to placental
deficiency. Proc Natl Acad Sci USA. 2007;104:6293–8. Article CAS PubMed PubMed Central Google Scholar * Takamoto N, Kurihara I, Lee K, Demayo FJ, Tsai MJ, Tsai SY. Haploinsufficiency of
chicken ovalbumin upstream promoter transcription factor II in female reproduction. Mol Endocrinol. 2005;19:2299–308. Article CAS PubMed Google Scholar * Takamoto N, You LR, Moses K,
Chiang C, Zimmer WE, Schwartz RJ, et al. COUP-TFII is essential for radial and anteroposterior patterning of the stomach. Development. 2005;132:2179–89. Article CAS PubMed Google Scholar
* You LR, Takamoto N, Yu CT, Tanaka T, Kodama T, Demayo FJ, et al. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc Natl Acad Sci USA.
2005;102:16351–6. Article CAS PubMed PubMed Central Google Scholar * Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum
quantified from variation in 141,456 humans. Nature. 2020;581:434–43. Article CAS PubMed PubMed Central Google Scholar * Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell
T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. Article CAS PubMed PubMed Central Google Scholar * Arsov T, Kelecic J, Frkovic SH,
Sestan M, Kifer N, Andrews D, et al. Expanding the clinical spectrum of pathogenic variation in NR2F2: asplenia. Eur J Med Genet. 2021;64:104347. Article CAS PubMed Google Scholar * Qiao
XH, Wang Q, Wang J, Liu XY, Xu YJ, Huang RT, et al. A novel NR2F2 loss-of-function mutation predisposes to congenital heart defect. Eur J Med Genet. 2018;61:197–203. Article PubMed Google
Scholar * Reuter MS, Chaturvedi RR, Liston E, Manshaei R, Aul RB, Bowdin S, et al. The Cardiac Genome Clinic: implementing genome sequencing in pediatric heart disease. Genet Med.
2020;22:1015–24. Article CAS PubMed PubMed Central Google Scholar * Richter F, Morton SU, Kim SW, Kitaygorodsky A, Wasson LK, Chen KM, et al. Genomic analyses implicate noncoding de
novo variants in congenital heart disease. Nat Genet. 2020;52:769–77. Article CAS PubMed PubMed Central Google Scholar * Wang J, Abhinav P, Xu YJ, Li RG, Zhang M, Qiu XB, et al. NR2F2
loss‑of‑function mutation is responsible for congenital bicuspid aortic valve. Int J Mol Med. 2019;43:1839–46. CAS PubMed Google Scholar * Upadia J, Gonzales PR, Robin NH. Novel de novo
pathogenic variant in the NR2F2 gene in a boy with congenital heart defect and dysmorphic features. Am J Med Genet A. 2018;176:1423–26. Article CAS PubMed Google Scholar * Kammoun M,
Souche E, Brady P, Ding J, Cosemans N, Gratacos E, et al. Genetic profile of isolated congenital diaphragmatic hernia revealed by targeted next-generation sequencing. Prenat Diagn.
2018;38:654–63. Article CAS PubMed Google Scholar * Bashamboo A, Eozenou C, Jorgensen A, Bignon-Topalovic J, Siffroi JP, Hyon C, et al. Loss of function of the nuclear receptor NR2F2,
encoding COUP-TF2, causes testis development and cardiac defects in 46,xx children. Am J Hum Genet. 2018;102:487–93. Article CAS PubMed PubMed Central Google Scholar * Qiao L, Wynn J,
Yu L, Hernan R, Zhou X, Duron V, et al. Likely damaging de novo variants in congenital diaphragmatic hernia patients are associated with worse clinical outcomes. Genet Med. 2020;22:2020–28.
Article CAS PubMed PubMed Central Google Scholar * Li AH, Hanchard NA, Furthner D, Fernbach S, Azamian M, Nicosia A, et al. Whole exome sequencing in 342 congenital cardiac left sided
lesion cases reveals extensive genetic heterogeneity and complex inheritance patterns. Genome Med. 2017;9:95. Article PubMed PubMed Central Google Scholar * High FA, Bhayani P, Wilson
JM, Bult CJ, Donahoe PK, Longoni M. De novo frameshift mutation in COUP-TFII (NR2F2) in human congenital diaphragmatic hernia. Am J Med Genet A. 2016;170:2457–61. Article CAS PubMed
PubMed Central Google Scholar * Matsunami N, Shanmugam H, Baird L, Stevens J, Byrne JL, Barnhart DC, et al. Germline but not somatic de novo mutations are common in human congenital
diaphragmatic hernia. Birth Defects Res. 2018;110:610–17. Article CAS PubMed PubMed Central Google Scholar * Schwab ME, Dong S, Lianoglou BR, Aguilar Lucero AF, Schwartz GB, Norton ME,
et al. Exome sequencing of fetuses with congenital diaphragmatic hernia supports a causal role for NR2F2, PTPN11, and WT1 variants. Am J Surg. 2022;223:182–86. Article PubMed Google
Scholar * Zidoune H, Ladjouze A, Chellat-Rezgoune D, Boukri A, Dib SA, Nouri N, et al. Novel genomic variants, atypical phenotypes and evidence of a digenic/oligogenic contribution to
disorders/differences of sex development in a large North African cohort. Front Genet. 2022;13:900574. Article CAS PubMed PubMed Central Google Scholar * Carvalheira G, Malinverni AM,
Moyses-Oliveira M, Ueta R, Cardili L, Monteagudo P, et al. The natural history of a man with ovotesticular 46,XX DSD caused by a novel 3-Mb 15q26.2 deletion containing NR2F2 gene. J Endocr
Soc. 2019;3:2107–13. Article PubMed PubMed Central Google Scholar * Brady PD, DeKoninck P, Fryns JP, Devriendt K, Deprest JA, Vermeesch JR. Identification of dosage-sensitive genes in
fetuses referred with severe isolated congenital diaphragmatic hernia. Prenat Diagn. 2013;33:1283–92. Article CAS PubMed Google Scholar * Poot M, Verrijn Stuart AA, van Daalen E, van
Iperen A, van Binsbergen E, Hochstenbach R. Variable behavioural phenotypes of patients with monosomies of 15q26 and a review of 16 cases. Eur J Med Genet. 2013;56:346–50. Article PubMed
Google Scholar * Mosca AL, Pinson L, Andrieux J, Copin H, Bigi N, Puechberty J, et al. Refining the critical region for congenital diaphragmatic hernia on chromosome 15q26 from the study of
four fetuses. Prenat Diagn. 2011;31:912–4. CAS PubMed Google Scholar * Dateki S, Fukami M, Tanaka Y, Sasaki G, Moriuchi H, Ogata T. Identification of chromosome 15q26 terminal deletion
with telomere sequences and its bearing on genotype-phenotype analysis. Endocr J. 2011;58:155–9. Article CAS PubMed Google Scholar * Rump P, Dijkhuizen T, Sikkema-Raddatz B, Lemmink HH,
Vos YJ, Verheij JB, et al. Drayer’s syndrome of mental retardation, microcephaly, short stature and absent phalanges is caused by a recurrent deletion of chromosome 15(q26.2—>qter). Clin
Genet. 2008;74:455–62. Article CAS PubMed Google Scholar * Davidsson J, Collin A, Bjorkhem G, Soller M. Array based characterization of a terminal deletion involving chromosome subband
15q26.2: an emerging syndrome associated with growth retardation, cardiac defects and developmental delay. BMC Med Genet. 2008;9:2. Article PubMed PubMed Central Google Scholar * Poot M,
Eleveld MJ, van 't Slot R, van Genderen MM, Verrijn Stuart AA, Hochstenbach R, et al. Proportional growth failure and oculocutaneous albinism in a girl with a 6.87 Mb deletion of
region 15q26.2—>qter. Eur J Med Genet. 2007;50:432–40. Article PubMed Google Scholar * Rujirabanjerd S, Suwannarat W, Sripo T, Dissaneevate P, Permsirivanich W, Limprasert P. De novo
subtelomeric deletion of 15q associated with satellite translocation in a child with developmental delay and severe growth retardation. Am J Med Genet A. 2007;143A:271–6. Article PubMed
Google Scholar * Hoang TT, Goldmuntz E, Roberts AE, Chung WK, Kline JK, Deanfield JE, et al. The congenital heart disease genetic network study: cohort description. PLoS One.
2018;13:e0191319. Article PubMed PubMed Central Google Scholar * Pediatric Cardiac Genomics C, Gelb B, Brueckner M, Chung W, Goldmuntz E, Kaltman J, et al. The congenital heart disease
genetic network study: rationale, design, and early results. Circ Res. 2013;112:698–706. Article Google Scholar * Rastetter RH, Bernard P, Palmer JS, Chassot AA, Chen H, Western PS, et al.
Marker genes identify three somatic cell types in the fetal mouse ovary. Dev Biol. 2014;394:242–52. Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We would like
to thank all the families who participated in this study. The National Institutes of Health (NIH) Common Fund, through the Office of Strategic Coordination and the Office of the NIH
Director, the Vanderbilt University Medical Center clinical site (U01HG007674). FUNDING The DDD study presents independent research commissioned by the Health Innovation Challenge Fund
[grant number HICF-1009-003], a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Sanger Institute [grant number WT098051]. The views
expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The study was approved by the UK Research Ethics
Committee (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for
Health Research, through the Comprehensive Clinical Research Network. Wendy Chung acknowledges funding support from grants P01HD068250 and U01 HL131003. Sara Sanz Benito acknowledges funding
from the Fondo de Investigación Sanitaria (PI15/01647 [SB-S]). Maria Francesca Bedeschi, Nuria C Bramswig and Ariane Schmetz are members of the European Reference Network on Rare Congenital
Malformations and Rare Intellectual Disability ERN-ITHACA [EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01-2016/739516]. Nuria C Bramswig and Ariane Schmetz acknowledge that this
work was partly done within the Zentrum für Seltene Erkrankungen of the University Hospital Düsseldorf (ZSED). AUTHOR INFORMATION Author notes * A full list of members and their affiliations
appears in the Supplementary Information. AUTHORS AND AFFILIATIONS * Department of Pathology & Cell Biology, Columbia University Irving Medical Center, New York, NY, USA Mythily
Ganapathi & Amanda Thomas-Wilson * Children’s Hospital of Philadelphia, Philadelphia, PA, USA Leticia S. Matsuoka, Michael March, Dong Li, Elaine Zackai & Elizabeth Bhoj * Vanderbilt
Genetics Institute, Vanderbilt University Medical Center, Nashville, TN, USA Elly Brokamp * CIBERER, ISCIII. Institute of Medical and Molecular Genetics (INGEMM), Disorder of Sex
Development Multidisciplinary Unit, Hospital Universitario La Paz, Madrid, Spain Sara Benito-Sanz * Victorian Clinical Genetics Service, Murdoch Children’s Research Institute, Melbourne,
VIC, Australia Susan M. White * Department of Pediatrics, University of Melbourne, Melbourne, VIC, Australia Susan M. White * Wessex Clinical Genetics Service, University Hospital
Southampton NHS Trust, Southampton, UK Katherine Lachlan * Department of Human Genetics and Genomic Medicine, Southampton University, Southampton, UK Katherine Lachlan * Department of
Pediatrics, Columbia University Irving Medical Center, New York, NY, USA Priyanka Ahimaz, Anshuman Sewda, John P. Schacht, Alejandro D. Iglesias & Wendy K. Chung * Department of
Biomedical Informatics, Vanderbilt University Medical Center, Nashville, TN, USA Lisa Bastarache * Division of Genetics and Genomics, Boston Children’s Hospital, Harvard Medical School,
Boston, MA, 02115, USA Joan M. Stoler & Stephanie Lucia * Institute of Human Genetics, Medical Faculty and University Hospital Düsseldorf, Heinrich Heine University Düsseldorf, 40225,
Düsseldorf, Germany Nuria C. Bramswig & Ariane Schmetz * Exeter Genomics Laboratory, Royal Devon & Exeter NHS Foundation Trust, Exeter, UK Julia Baptista & Karen Stals *
Peninsula Medical School, Faculty of Health, University of Plymouth, PL4 8AA, Plymouth, UK Julia Baptista * Service de Génétique CH Bretagne Atlantique-Vannes, Vannes, France Florence
Demurger * Nantes Université, CHU de Nantes, CNRS, INSERM, l’institut du thorax, F-44000, Nantes, France Benjamin Cogne & Bertrand Isidor * Nantes Université, CHU de Nantes, Service de
Génétique médicale, F-44000, Nantes, France Benjamin Cogne & Bertrand Isidor * Medical Genetics Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy Maria
Francesca Bedeschi * Medical Genetics, ASST Santi Paolo e Carlo, San Paolo Hospital, Università degli Studi di Milano, Milan, Italy Angela Peron * Division of Medical Genetics, Department of
Pediatrics, University of Utah, Salt Lake City, UT, USA Angela Peron * INSERM UMR1163, Institut Imagine, Université Paris-Cité, Paris, France Jeanne Amiel & Christopher T. Gordon *
Service de Médecine Génomique des Maladies Rares, Hôpital Necker-Enfants Malades, AP-HP, Paris, France Jeanne Amiel * West Midlands Regional Clinical Genetics Service and Birmingham Health
Partners, Birmingham Women’s and Children’s Hospitals NHS Foundation Trust, Birmingham, UK Jenny Morton * Clinical Genetics, Department of Pediatrics, Gregorio Marañón General University
Hospital, Madrid, Spain Verónica Seidel * Department of Pediatrics, Baylor College of Medicine, San Antonio, TX, USA Stephanie M. Baskin * Department of Molecular and Human Genetics, Baylor
College of Medicine, Houston, TX, USA Stephanie M. Baskin * Genomic Medicine Center, Children’s Mercy Hospital, Kansas City, MO, USA Isabelle Thiffault * Department of Pediatrics, Vanderbilt
University Medical Center, Nashville, TN, USA Joy D. Cogan * Department of Clinical Genetics, Addenbrooke’s Hospital, Cambridge University Hospitals NHS, Foundation Trust, Cambridge, UK
Sarah Bowdin * Vanderbilt University Medical Center, Division of Medical Genetics and Genomic Medicine, 1161 21st Ave. S, DD-2205 Medical Center North, Nashville, TN, 37232, USA Joy D. Cogan
Authors * Mythily Ganapathi View author publications You can also search for this author inPubMed Google Scholar * Leticia S. Matsuoka View author publications You can also search for this
author inPubMed Google Scholar * Michael March View author publications You can also search for this author inPubMed Google Scholar * Dong Li View author publications You can also search for
this author inPubMed Google Scholar * Elly Brokamp View author publications You can also search for this author inPubMed Google Scholar * Sara Benito-Sanz View author publications You can
also search for this author inPubMed Google Scholar * Susan M. White View author publications You can also search for this author inPubMed Google Scholar * Katherine Lachlan View author
publications You can also search for this author inPubMed Google Scholar * Priyanka Ahimaz View author publications You can also search for this author inPubMed Google Scholar * Anshuman
Sewda View author publications You can also search for this author inPubMed Google Scholar * Lisa Bastarache View author publications You can also search for this author inPubMed Google
Scholar * Amanda Thomas-Wilson View author publications You can also search for this author inPubMed Google Scholar * Joan M. Stoler View author publications You can also search for this
author inPubMed Google Scholar * Nuria C. Bramswig View author publications You can also search for this author inPubMed Google Scholar * Julia Baptista View author publications You can also
search for this author inPubMed Google Scholar * Karen Stals View author publications You can also search for this author inPubMed Google Scholar * Florence Demurger View author
publications You can also search for this author inPubMed Google Scholar * Benjamin Cogne View author publications You can also search for this author inPubMed Google Scholar * Bertrand
Isidor View author publications You can also search for this author inPubMed Google Scholar * Maria Francesca Bedeschi View author publications You can also search for this author inPubMed
Google Scholar * Angela Peron View author publications You can also search for this author inPubMed Google Scholar * Jeanne Amiel View author publications You can also search for this author
inPubMed Google Scholar * Elaine Zackai View author publications You can also search for this author inPubMed Google Scholar * John P. Schacht View author publications You can also search
for this author inPubMed Google Scholar * Alejandro D. Iglesias View author publications You can also search for this author inPubMed Google Scholar * Jenny Morton View author publications
You can also search for this author inPubMed Google Scholar * Ariane Schmetz View author publications You can also search for this author inPubMed Google Scholar * Verónica Seidel View
author publications You can also search for this author inPubMed Google Scholar * Stephanie Lucia View author publications You can also search for this author inPubMed Google Scholar *
Stephanie M. Baskin View author publications You can also search for this author inPubMed Google Scholar * Isabelle Thiffault View author publications You can also search for this author
inPubMed Google Scholar * Joy D. Cogan View author publications You can also search for this author inPubMed Google Scholar * Christopher T. Gordon View author publications You can also
search for this author inPubMed Google Scholar * Wendy K. Chung View author publications You can also search for this author inPubMed Google Scholar * Sarah Bowdin View author publications
You can also search for this author inPubMed Google Scholar * Elizabeth Bhoj View author publications You can also search for this author inPubMed Google Scholar CONSORTIA UNDIAGNOSED
DISEASES NETWORK * Joy D. Cogan CONTRIBUTIONS MG collected, analyzed and interpreted the data, drafted the introduction, results, discussion, figures, and tables. MG, LSM, MM, DL, EB, SBS,
SMW, KL, PA, AS, LB, ATW, JMS, KS, FD, BC, BI, MFB, AP, JA, EZ, JPS, ADI, JM, VS, SL, SB, IT, JDC, CTG, WKC, SB, EB, NCB, AS provided the clinical data, wrote the clinical case descriptions,
methods, critically reviewed and edited the manuscript. EB conceived the study, interpreted the data, and critically reviewed the manuscript. CORRESPONDING AUTHOR Correspondence to
Elizabeth Bhoj. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICAL APPROVAL The Institutional Review Board of the Children’s Hospital of Philadelphia
approved this study. Informed consent was obtained from all individual participants included in the study. Families of individuals 2 &12 consented for publication of images. ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
INFORMATION SUPPLEMENTARY FIGURES SUPPLEMENTARY TABLE 1 SUPPLEMENTARY TABLE 2 SUPPLEMENTARY TABLE 3 SUPPLEMENTARY TABLE 4 RIGHTS AND PERMISSIONS Springer Nature or its licensor (e.g. a
society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript
version of this article is solely governed by the terms of such publishing agreement and applicable law. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ganapathi, M.,
Matsuoka, L.S., March, M. _et al._ Heterozygous rare variants in _NR2F2_ cause a recognizable multiple congenital anomaly syndrome with developmental delays. _Eur J Hum Genet_ 31, 1117–1124
(2023). https://doi.org/10.1038/s41431-023-01434-5 Download citation * Received: 08 February 2023 * Revised: 07 June 2023 * Accepted: 11 July 2023 * Published: 27 July 2023 * Issue Date:
October 2023 * DOI: https://doi.org/10.1038/s41431-023-01434-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative