The genetic landscape of polycystic kidney disease in Ireland
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Polycystic kidney diseases (PKDs) comprise the most common Mendelian forms of renal disease. It is characterised by the development of fluid-filled renal cysts, causing progressive loss of
kidney function, culminating in the need for renal replacement therapy or kidney transplant. Ireland represents a valuable region for the genetic study of PKD, as family sizes are
traditionally large and the population relatively homogenous. Studying a cohort of 169 patients, we describe the genetic landscape of PKD in Ireland for the first time, compare the clinical
features of patients with and without a molecular diagnosis and correlate disease severity with autosomal dominant pathogenic variant type. Using a combination of molecular genetic tools,
including targeted next-generation sequencing, we report diagnostic rates of 71–83% in Irish PKD patients, depending on which variant classification guidelines are used (ACMG or Mayo clinic
respectively). We have catalogued a spectrum of Irish autosomal dominant PKD pathogenic variants including 36 novel variants. We illustrate how apparently unrelated individuals carrying the
same autosomal dominant pathogenic variant are highly likely to have inherited that variant from a common ancestor. We highlight issues surrounding the implementation of the ACMG guidelines
for variant pathogenicity interpretation in PKD, which have important implications for clinical genetics.
Polycystic kidney diseases (PKDs) are the most common forms of the inherited renal disease affecting an estimated 12.5 million individuals worldwide [1]. PKD is a leading cause of end-stage
kidney disease (ESKD) and the prevalence of PKD related ESKD in Europe ranges from 30 per million population (pmp) in Switzerland to greater than 180 pmp in regions of Spain [2]. In Northern
Ireland the prevalence of ESKD due to PKD is reported as 115 pmp [2].
PKD can be autosomal dominant, autosomal recessive (ADPKD and ARPKD) or syndromic and can manifest prior to birth on prenatal ultrasound or as late as the eighth decade of life. Most ADPKD
patients (85%) reach ESKD by age 65 years [3]. Approximately 90% of ADPKD cases have an identifiable pathogenic genetic variant in PKD1 or PKD2; leaving ~10% with an unknown genetic cause
[4]. PKD1 and PKD2 account for around 80–85% and 15–20% of cases with a molecular diagnosis, respectively [5,6,7,8,9,10]. Copy number changes in the form of insertions and deletions over 1
kb in size [11] within PKD1 and PKD2 are responsible for 1–5% of cases [5, 8, 12, 13]. Additional genes account for rarer forms of ADPKD (18 years,
Did not have a previous genetic diagnosis of PKD (i.e. causal AR/AD pathogenic variant was unknown),
Had a family history of PKD defined by the presence of a second-degree relative or closer, with the disease, at the time of recruitment to the study,
Met the unified criteria for ultrasonography diagnosis of ADPKD [27],
Had no family history of PKD but evidence of >5 cysts on ultrasound examination and no evidence of an alternate cystic kidney disease.
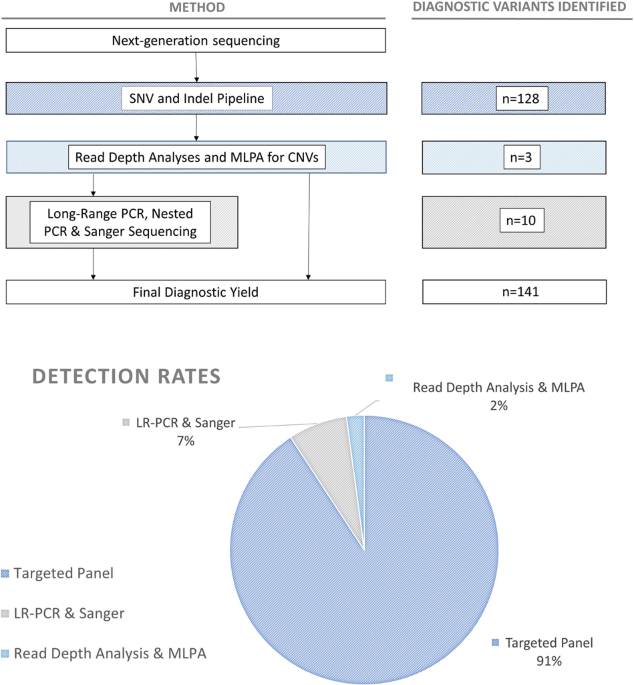
NGS was performed on 155 samples using a custom targeted panel (Roche SeqCap EZ Choice) of 227 known renal disease genes (99.9% of target bases covered, gene list previously published [28])
on an Illumina Platform (MiSeq or NextSeq). Fourteen samples were sequenced using an alternative panel covering 11 genes (Roche HeatSeq gene list provided in Supplementary Table 1).
Bioinformatic analyses were conducted using a GATK Best Practices based pipeline [29]. All variants discussed in this manuscript have been submitted to ClinVar (see Supplementary Table 3)
and requests for access to raw sequence data can be made via communication with the corresponding author.
Candidate AD and AR pathogenic variants were identified as those that were: (1) in established renal disease genes, (2) within exonic and splicing regions (3) predicted damaging by either
PolyPhen [30] or SIFT [31] and (4) with a minor allele frequency (MAF) (as per gnomAD [32]) of 5%, or MAF A:p.(Arg3750Gln)) has an allele frequency in gnomAD of 4 × 10−6 and has been
reported in four families in the PKDB mutation database (https://pkdb.mayo.edu/index.html), suggesting that it is a relatively common and thus probably older PKD variant. Alternatively,
these variants may represent independent mutation events. This type of genetic distance analysis may be of use when assigning pathogenicity to a variant using ACMG guidelines. For example,
the variant carried by families F391 and F399 could be reclassified using ACMG guidelines from a VUS to likely pathogenic as a result of the additional segregation evidence provided by this
relationship analysis.
We have replicated previous reports [16] that link PKD1/2 AD pathogenic variants with disease severity and the association between AD pathogenic variant type (PKD1 ‘truncating’, PKD1
‘non-truncating’ or PKD2) and age at ESKD [17]. This association did not extend to more finely categorised variant types, perhaps as a result of low numbers in these smaller categories. We
did not observe the previously reported [41] difference in the age at which patients with PKD2 truncating or PKD2 non-truncating AD pathogenic variants reach ESKD, probably due to lack of
study power. These results are consistent with previous reports [17, 18]. However, previous reports have indicated a difference in levels of eGFR between these groups [18]. Our
screen-negative patients appeared to have similar rates of renal survival to those with AD PKD1 non-truncating pathogenic variants and had significantly lower renal survival than those with
PKD2 AD pathogenic variants.
We observed three individuals who carried additional variants that satisfied the ACMG guidelines for variant pathogenicity (‘likely pathogenic’/’pathogenic’) (Supplementary Table 8). It is
unclear based on the patient phenotypes available whether these variants may have an impact on the severity of the disease in these cases. Two of these patients carried the same splicing
variant in COL4A4 (c.2164 + 2T > G). Collagen gene variants including those in COL4A4 are traditionally associated with Alport syndrome/thin basement membrane disease but have been reported
previously in-tandem with a PKD AD pathogenic variant and may have a modifier effect on primary PKD variants [42]. The main feature of COL4A4-linked disease is haematuria and progressive
renal failure associated with progressive hearing loss. There was no evidence of hearing loss in either pedigree.
Whilst the ACMG guidelines have been widely embraced by the scientific community, it is recognised that there are deficiencies. These guidelines perform best for paediatric-onset rare
diseases. For adult-onset disorders such as PKD and those with reduced penetrance, there are a number of weaknesses:
Missense variants are common and score as only moderate evidence for pathogenicity while truncating variants score as strong evidence. This increases the likelihood that missense variants
will be scored as a VUS.
Population databases such as gnomAD are used to estimate the frequency of variants in the healthy population. This is a reasonable approach for paediatric rare diseases where penetrance is
almost 100% by adulthood. However, it is possible for PKD that a small number of affected patients may be included in the ‘healthy’ population before presenting with symptoms. This may lead
an AD pathogenic variant to be deemed as benign/VUS.
The incidence of de novo variants is lower in PKD than in severe paediatric-onset diseases. If a variant is de novo this can be used as strong evidence of pathogenicity within ACMG. As most
PKD is inherited, it means that many more variants remain as a VUS.
In diseases like PKD in which there is extreme allelic heterogeneity, it is less likely that a variant will have been previously reported in a disease database, reducing the opportunity to
assign strong evidence on the basis of previously established pathogenicity.
We assessed the utility of the ACMG guidelines for clinical variant pathogenicity interpretation in PKD and compared these guidelines to those widely used in PKD research from the Mayo
Clinic. An important difference between these tools is that the ACMG guidelines were developed for clinical genetics use, while the guidelines from the Mayo Clinic were designed for research
use only. Our results highlight an issue surrounding the use of the ACMG guidelines, with regard to missense PKD1 variant assessment. These missense variants are difficult to classify as
likely pathogenic or pathogenic using the ACMG guidelines unless they have been extensively studied and reported previously [43], largely due to the variability in regional pathogenicity of
PKD1 [10]. While it is imperative that pathogenicity guidelines are conservative to control for ‘over-calling’ of Pathogenic/Likely Pathogenic variants, the conservative nature of the ACMG
guidelines can be problematic as novel PKD1 variants are common, with 31% of diagnostic variants identified being a novel finding from our cohort. Equally, it is important to note that
higher diagnostic rates obtained when using the Mayo Clinic research classification tool may be enriched for false-positives, which in a clinical setting could cause harm to patients. The
2015 ACMG guidelines refer to the development of focused guidance for the classification of variants in specific genes since pathogenicity criteria vary by gene and disease [44]. Indeed,
gene-specific guidelines for the interpretation of variants have been proposed for other diseases [23, 45,46,47]. These disease-specific modifications have in some cases increased the number
of variants classified as VUS and are not necessarily more permissive of missense variants [45]. We suggest that a tailored set of ACMG-based guidelines may facilitate the standardisation
of interpretation of variants in PKD1.
Studies of PKD in US, French, Italian and German populations have contributed to the development of a catalogue of PKD AR and AD pathogenic variants [5, 12, 48,49,50,51]. However, the
broader European population (and non-Caucasian populations) has not been studied in as much depth as the US one [17, 50, 52]. There have been limited studies cataloguing the spectrum of AD
and AR pathogenic variants in the UK and Ireland to date [16], although the inclusion of cystic renal disease patients in the Genomics England initiative will likely result in a catalogue of
UK AD and AR pathogenic variants.
There were a number of limitations to this study. The coverage achieved for PKD1 (NG_008617.1) exon 1 was not sufficient for variant identification, necessitating gap-filling Sanger
sequencing in patients without a molecular diagnosis from NGS. It is possible that due to low coverage of this region, or due to pseudogene homology to PKD1 that some patients carried
additional, clinically relevant variants that were not detected. The sequencing methods used in this study were also unable to detect small insertions and deletions in the variable number
tandem repeat region of MUC1.
A genetic diagnosis not only informs on prognosis, but facilitates cascade testing of relatives considering donating a kidney to a family member, and provides information when planning a
family or making decisions about care. A confirmed genetic diagnosis opens the possibility of pre-implantation screening for couples with a family history of PKD and may ensure that patients
receive follow-up testing for common PKD complications including cerebral aneurysms and liver cysts. We recommend that PKD testing adopts a combination of approaches for the molecular
diagnosis of PKD patients. Finally, we recommend further standardisation of variant pathogenicity assessment within the PKD community, but highlight some potential issues with the
implementation of established ACMG pathogenicity guidelines which may require some modification for use in PKD.
KB is supported by an Enterprise Partnership Scheme Fellowship Award (2019) from The Irish Research Council, in conjunction with Punchestown Kidney Research Fund (EPSPD/2019/213). The
authors also acknowledge funding received from the Beaumont Hospital Foundation, the Royal Irish Academy and the Royal College of Surgeons in Ireland. SC is supported by the Irish Clinical
Academic Training (ICAT) Programme, supported by the Wellcome Trust and the Health Research Board (Grant Number 203930/B/16/Z), the Health Service Executive National Doctors Training and
Planning and the Health and Social Care, Research and Development Division, Northern Ireland. We also acknowledge that this work would not be possible without the participation of the
consenting patients and their families.
These authors contributed equally: Gianpiero L. Cavalleri, Peter Conlon
School of Pharmacy and Biomolecular Science, Royal College of Surgeons in Ireland, Dublin, Ireland
Katherine A. Benson, Edmund Gilbert, Sarah Khamis, Robert Carton, Gianpiero L. Cavalleri & Peter Conlon
Department of Nephrology, Beaumont Hospital, Dublin, Ireland
Susan L. Murray, Elhussein Elhassan, Eoin T. Conlon, Claire Kennedy, Shane Conlon, Dervla Connaughton & Peter Conlon
Division of Nephrology and Hypertension, Mayo Clinic, Rochester, MN, USA
Department of Renal Medicine, University Hospital Limerick, Limerick, Ireland
Regenerative Medicine Institute (REMEDI) at CÚRAM Centre for Research in Medical Devices, School of Medicine, National University of Ireland Galway, Galway, Ireland
Nephrology Department, Galway University Hospitals, Saolta University Healthcare Group, Galway, Ireland
Division of Intramural Research, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA
Department of Clinical Genetics, Children’s University Hospital, Temple Street, Dublin, Ireland
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Table 3: Supplementary Table 3: Genetic Diagnosis in patients in which a disease-causing variant of PKD was identified
Supplementary Table 11: Genetic scoring of nontruncating PKD1 and PKD2 variants scored as VUS by the ACMG classification employing the Mayo Research Mutation Classification Algorithm
Anyone you share the following link with will be able to read this content: