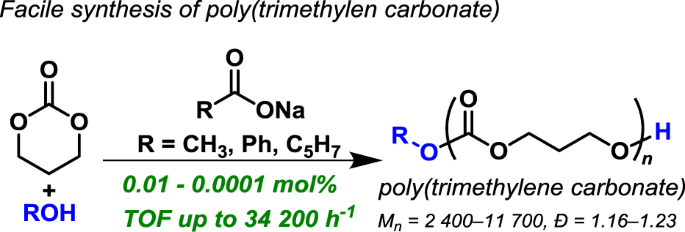
Facile synthesis of poly(trimethylene carbonate) by alkali metal carboxylate-catalyzed ring-opening polymerization
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Alkali metal carboxylates, including sodium acetate, sodium benzoate, and sodium sorbate, which are all readily available and widely used as food additives, were found to promote
the ring-opening polymerization (ROP) of trimethylene carbonate (TMC) to produce poly(trimethylene carbonate) (PTMC). The sodium acetate-catalyzed ROP of TMC proceeded in the presence of an
alcohol initiator under solvent-free conditions at 70 °C, even at very low catalyst loadings of 0.01–0.0001 mol%. The controlled nature of this ROP system enabled the synthesis of PTMCs with
predicted molecular weights ranging from 2400 to 11 700 g mol−1 and narrow dispersities (~1.23). Importantly, ROP is initiated by an alcohol initiator, allowing PTMC production with desired
functional groups, such as azido, alkyne, and methacrylate groups, at the _α_-chain end. Furthermore, the poly(l-lactic acid)-_b_-PTMC-_b_-poly(l-lactic acid) triblock copolymer, a
biodegradable thermoplastic elastomer, was successfully synthesized in one pot via the sodium acetate-catalyzed ring-opening block copolymerization of TMC and l-lactide with a
1,3-propanediol initiator. You have full access to this article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS CHEMOSELECTIVE TRANSESTERIFICATION AND POLYMER
SYNTHESIS USING A ZINCATE COMPLEX Article 18 September 2020 SYNTHESIS AND CHARACTERIZATION OF ESTER-FREE POLY(TRIMETHYLENE CARBONATE) BEARING LONG-ALKYL MOIETIES AND ITS DEGRADATION Article
15 November 2024 DOUBLE-CYCLOPOLYMERIZATION USING TRIFUNCTIONAL INCOMPLETELY CONDENSED CAGE SILSESQUIOXANE WITH METHACRYLOYL GROUPS Article 12 December 2022 INTRODUCTION Aliphatic
polycarbonates (APCs) have attracted significant attention for their applications in biomedical fields due to their excellent biocompatibility and biodegradability [1,2,3,4]. APCs are better
suited for biomedical applications than are aliphatic polyesters such as poly(lactic acid), poly(glycolic acid), and poly(caprolactone), as aliphatic polyesters generate acidic compounds
during degradation, which may affect tissues or DNA [4,5,6]. In addition, the hydrolytic degradation of APCs is extremely slow, but these molecules can be rapidly degraded in vivo [7].
Because of these advantages, APCs have been recently studied as components of drug delivery carriers and scaffolds for tissue engineering [1,2,3,4]. In addition to their biomedical
applications, APCs can be used as soft segments of polyurethanes, resulting in large elongations at break and high tensile strengths while maintaining their good biocompatibility [8]. Common
synthetic approaches for APC production can be classified into three categories: polycondensation of aliphatic diols with phosgene or dialkyl carbonates, copolymerization of epoxides with
CO2, and ring-opening polymerization (ROP) of cyclic carbonates. Industrially important APC diols are mainly synthesized using the polycondensation approach, but the molecular weight and
dispersity are difficult to control. In contrast, the ROP approach is highly preferable for controlling the molecular weight, dispersity, and chain-end groups. Thus, significant efforts have
been dedicated to the development of novel catalysts for the ROP of cyclic carbonates. ROP of cyclic carbonates is conventionally performed using an organometallic catalyst, such as
stannous octoate [9, 10], dibutyltin dioctanoate [11], or aluminum alkoxides [12]. Alternatively, organocatalytic ROPs of cyclic carbonates have recently been developed to achieve metal free
and environmentally benign APC synthesis. The first study of the organocatalytic ROP of cyclic carbonate was reported by Waymouth and Hedrick in 2007 using amidine, guanidine,
_N_-heterocyclic carbene, and thiourea/amine [13]. These catalysts enabled ROP to afford well-defined APCs under mild conditions. Later, organic bases and acids, as well as bifunctional
acid/base systems, were developed. Strong organic acids, such as methanesulfonic acid and triflic acid, are representative examples of acidic catalysts [14]. The bifunctional acid/base
catalyst includes thiourea/amine [13] and trifluoroacetic acid/7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene systems [15]. Our group also found that diphenyl phosphate (DPP) [16] and
trimethyl glycine [17] can form a good catalyst system for the ROP of cyclic carbonates. Notably, the DPP-catalyzed ROP of trimethylene carbonate (TMC) proceeded under solvent-free
conditions even at a low catalyst loading of 0.1 mol% [18]. Although significant effort has been devoted to developing novel organocatalysts, an industrially feasible catalyst that is
inexpensive, easy to handle, and safe remains elusive. Many of the organocatalytic ROP systems for cyclic carbonates require relatively high catalyst loadings of 0.1–5 mol%, but to meet
industrial requirements, the catalyst loading must be further reduced. Recently, we discovered the catalytic ability of alkali metal carboxylates for the ROP of lactides, affording
well-defined and narrowly dispersed polylactides [19]. Alkali metal carboxylates, including sodium acetate (CH3COONa) and sodium benzoate (PhCOONa), are widely used as food additives,
meeting the industrial requirement of low cost, easy handling, and safety. Furthermore, a preliminary study revealed that CH3COONa catalyzes the ROP of TMC, affording poly(trimethylene
carbonate) (PTMC) with a narrow dispersity. To explore the potential of this catalytic system for APC synthesis, herein, we investigated the ROP of TMC under solvent-free conditions using
the alkali metal carboxylates CH3COONa, PhCOONa, and sodium sorbate (C5H7COONa), which are all readily available and used as food additives (Scheme 1). The best catalytic activity was
observed using CH3COONa, which smoothly catalyzed the ROP of TMC even at a catalyst loading of 0.001 mol%. Furthermore, the catalyst system enabled the synthesis of narrowly dispersed PTMCs
with various molecular weights and desired chain-end functionalities. RESULTS AND DISCUSSION RING-OPENING POLYMERIZATION OF TRIMETHYLENE CARBONATE USING SODIUM CARBOXYLATES AS CATALYSTS We
initially investigated the ROP of TMC using different alkali metal carboxylates as catalysts, i.e., CH3COONa (run 1), sodium benzoate (PhCOONa; run 2), and sodium sorbate (C5H7COONa; run 3),
under solvent-free conditions (Table 1). ROP was conducted at 70 °C in the presence of 3-phenyl-1-propanol (PPA) as an initiator and with a catalyst loading of 0.01 mol%
([TMC]0/[PPA]0/[CH3COONa]0 ratio of 50/1/0.005). When CH3COONa was used as the catalyst, the monomer conversion reached 72.5% in 28 min of polymerization to afford PTMC, indicating
sufficient catalytic activity of CH3COONa for the ROP of TMC even at a catalyst loading of 0.01 mol% (run 1, Table 1). End group analysis of the obtained PTMC by 1H NMR revealed an _M_n,NMR
of value 4170 g mol−1, which agreed with the theoretical value (_M_n,th.) of 3 840 g mol−1. The size exclusion chromatography (SEC) trace exhibited a unimodal and narrow elution peak with a
dispersity (_Ð_) value of 1.17. These results indicated that the CH3COONa-catalyzed ROP of TMC proceeded in a well-controlled manner. When PhCOONa and C5H7COONa were used as catalysts, the
monomer conversion reached 69.6% in 38 min and 78.8% in 140 min (runs 2 and 3, respectively, in Table 1). Although each polymerization afforded a PTMC with a narrow _Ð_ value of 1.16, the
catalytic ability of PhCOONa and C5H7COONa was significantly less than that of CH3COONa, with a turnover frequency (TOF) of 11 000 h−1 for PhCOONa and 3 380 h−1 for C5H7COONa compared with a
TOF of 15 500 h−1 for CH3COONa. To further reduce the catalyst loading, ROP of TMC was performed using CH3COONa with catalyst loadings of 0.001 and 0.0001 mol% with respect to the monomer
([TMC]0/[PPA]0 = 50/1 at 70 °C; runs 4 and 5, respectively; see Table 1). In both cases, the monomer conversion reached ~70% in a reasonable polymerization time, affording PTMCs with narrow
_Ð_ values of 1.16–1.18. To exclude the possibility that trace impurities in the monomer catalyzed the ROP, we performed ROP in the absence of a catalyst at 70 °C (run 6 in Table 1). After
reacting for 130 min (same as run 4), the monomer conversion was 6.4%, and a low-molecular-weight oligomer was obtained. Thus, CH3COONa acted as the catalyst for the ROP of TMC. It should be
noted that typical organocatalysts for TMC polymerization require relatively high catalyst loadings: diphenyl phosphate (DPP; solvent-free) [18], trimethyl glycine (TMG; solvent-free) [17],
and 1,5,7-triazabicyclo-[4.4.0]dec-5-ene (TBD; in CH2Cl2) [13] required 0.1, 0.2, and 1 mol%, respectively, when the initial [TMC]0/[initiator]0 ratio was set to 50/1. Moreover, the highest
TOF of 34 200 h−1 was observed when using 0.001 mol% of CH3COONa, which was considerably higher than those of the other organocatalysts, e.g., DPP (TOF = 54 h−1 at 80 °C, solvent-free), TMG
(TOF = 750 h−1 at 70 °C, solvent-free), and TBD (TOF = 400 h−1 at r.t. in CH2Cl2). Next, to control the molecular weight, the ROP of TMC was conducted with various initial
monomer-to-initiator ratios ([TMC]0/[PPA]0 = 25/1, 100/1, and 200/1) at a catalyst loading of 0.001 mol% (runs 7–9 in Table 1, Fig. S1). For the ROP of TMC with an initial [TMC]0/[PPA]0
ratio of 25/1, the obtained PTMC showed an _M_n,NMR value of 2400 g mol−1 and a narrow _Ð_ value of 1.19. Similarly, ROP with initial [TMC]0/[PPA]0 ratios of 100/1 and 200/1 afforded PTMCs
with higher _M_n,NMR values of 8200 and 11 700 g mol−1 with narrow _Ð_ values of 1.23 and 1.12, respectively. When targeting high-molecular-weight PTMCs, the high molecular weight shoulder
in the SEC traces became pronounced because the propagation reaction competed with the intermolecular transesterification reactions (Fig. S1). STRUCTURAL ANALYSIS OF PTMC To obtain further
insight into the polymerization reaction, the chemical structure of the obtained PTMC was investigated by 1H NMR and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF)
mass spectral analyses. The 1H NMR spectrum of the PTMC obtained from run 4 showed signals corresponding to the PTMC main chain as well as minor signals arising from the PPA residue.
Importantly, the peak area of the PPA methylene proton (_a_ in Fig. 1) agreed well with that of the _ω_-chain end methylene proton (_g_ in Fig. 1), strongly suggesting that the
polymerization was initiated by PPA. It is well known that the ROP of cyclic carbonates often suffers from decarboxylation reactions, resulting in the formation of unwanted ether linkages on
the PTMC main chain [20]. However, no evidence of the ether linkage was observed for the PTMCs produced with CH3COONa. The mildly basic nature of the catalyst is likely responsible for the
clean polymerization. The MALDI-TOF mass spectral analysis provided further detailed structural information, especially in terms of end group structures of the PTMC. In the MALDI-TOF mass
spectrum (Fig. 2), a major series of peaks was observed in the range of 1500–5000 Da along with three minor series of peaks. The distance between each peak was 102.1 Da, corresponding to the
mass of the TMC repeating unit (102.0 Da). The main series of peaks, which is denoted by the filled circles (●), and the minor series of peaks denoted by open circles (○) were assigned to
the expected PTMC structure containing a PPA residue at the _α_-chain end and a hydroxyl group at the _ω_-chain end. For example, the peak observed at _m_/_z_ 3526.1 was assigned to the
33-mer PTMC containing the expected end group structures (calculated [M + Na]+ = 3527.1). In contrast, the minor series of peaks denoted by the filled squares (■) was assigned to PTMC
containing a 1,3-propanediol residue instead of a PPA residue. The minor series of peaks denoted by open squares (□) was assigned to the PTMC containing PPA residues at both chain ends.
These species were generated via an intermolecular transesterification reaction during polymerization (Fig. S2). INVESTIGATION INTO THE POLYMERIZATION PROPERTIES To obtain further insight
into the polymerization properties, we investigated the time course of the _M_n,SEC and _Ð_ values of the PTMCs obtained via ROP with a [TMC]0/[PPA]0/[CH3COONa]0 ratio of 50/1/0.005 at 70 °C
(Fig. 3(a)). The _M_n,SEC value linearly increased with increasing monomer conversion, suggesting the absence of chain transfer reactions. The _Ð_ value remained narrow (~1.26) throughout
the polymerization, except during the early stages. Moreover, the SEC elution peak of the resulting polymers retained a unimodal shape regardless of monomer conversion (Fig. 3(b)). When the
conversion reached 96.6%, a shoulder peak in the high-molecular-weight region became prominent. This suggests that the polymerization reaction competes with intermolecular
transesterification during the later stages of polymerization (conv. > 75%). Living polymerization reactions are known to follow a first-order kinetic model. However, the ROP system
investigated herein does not fit with the first-order kinetic model (Fig. S3(a)). The time versus conversion plot indicated that the propagation rate increased with increasing monomer
conversion (Fig. S3(b)). This could be explained by the insufficient solubility of the catalyst in the reaction medium. The catalyst gradually dissolved in the reaction medium during
polymerization, resulting in an increased propagation rate during the late stages of polymerization. Although this ROP system does not follow the first-order kinetic model, the _M_n,SEC of
the resulting PTMCs linearly increases with the monomer conversion. while a narrow _Ð_ value is retained, indicating the well-controlled nature of the polymerization. EFFECTS OF THE
COUNTERCATION ON CATALYTIC PERFORMANCE We previously reported that alkali metal carboxylates catalyzed the ROP of l-lactide (l-LA) via a dual activation mechanism involving hydroxyl chain
end/initiator activation and monomer activation [19]. The change in the alkali metal cation largely affected the TOF of the l-LA polymerization because the countercation participated in
monomer carbonyl group activation. However, no significant countercation effect was observed in the ROP of TMC using CH3COONa, potassium acetate (CH3COOK), or cesium acetate (CH3COOCs; Table
S1). In addition, the TOF was not affected by the countercation (34 200 h−1 for CH3COONa, 29 100 h−1 for CH3COOK, and 33 000 h−1 for CH3COOCs). This suggests that, for the ROP of TMC, the
countercations of the catalyst do not participate in monomer activation. Given that the carbonate group exhibits an intrinsically high dielectric property, the countercation should be well
solvated in the polymerization medium, allowing the carboxylate anion to be naked. The naked carboxylate anion activates the hydroxyl chain end/initiator as a base, which is responsible for
the extraordinarily high catalytic ability for the ROP of TMC. APPLICATION TO THE SYNTHESIS OF FUNCTIONALIZED APCS To demonstrate the applicability of the developed catalytic ROP system for
the synthesis of high value-added APCs, we examined the ROP of TMC using various functional initiators, including 6-azido-1-hexanol (AHA), 4-ethynylbenzyl alcohol (EBM), and 2-hydroxyethyl
methacrylate (HEMA; Table 2). AHA and EBM are functional initiators containing azido and alkyne groups, respectively, which can provide “clickable” PTMCs for constructing block copolymers.
The ROP with HEMA provided a PTMC macromonomer with a polymerizable methacrylate group. The ROP with functional initiators was conducted at an initial [TMC]0/[initiator]0/[CH3COONa] ratio of
50/1/0.0005, producing PTMCs with predictable molecular weights and narrow _Ð_ values. For example, the ROP with AHA reached 77.1% monomer conversion in 95 min, and the obtained PTMC
exhibited an _M_n,NMR value of 4080 g mol−1 and a narrow _Ð_ value of 1.18 (run 10 in Table 2). The 1H NMR spectrum of the obtained PTMCs showed a minor signal at 3.27 ppm arising from the
methylene adjacent to the azido group along with the major characteristic PTMC signals (Fig. S4). The PTMCs obtained using EBM and HEMA also showed 1H NMR signals arising from the
corresponding initiator residue, confirming that the ROP reactions were initiated by the functional initiators and afforded the corresponding _α_-functionalized PTMCs (Figs. S5 and S6).
Next, 1,3-propanediol (PD) was used as a difunctional initiator for the ROP of TMC to obtain PTMC-diol, which can be used as a soft segment for thermoplastic elastomers. The ROP with PD
yielded the desired PTMC-diol with a narrow _Ð_ value of 1.13 (run 13 in Table 2 and Fig. S7), which encouraged us to attempt the one-pot synthesis of a thermoplastic elastomer via
CH3COONa-catalyzed ROP. Thus, the synthesis of a poly(l-lactic acid)-_b_-PTMC-_b_-poly(l-lactic acid) (PLLA-_b_-PTMC-_b_-PLLA) triblock copolymer was performed (Fig. 4(a)), and this material
has been described as a biodegradable thermoplastic elastomer by Kim et al. [21]. After the ROP of TMC at an initial [TMC]0/[PD]0/[CH3COONa] ratio of 50/1/0.0005 at 70 °C for 120 min (conv.
= 87.1%), l-lactide (l-LA) and additional CH3COONa were added at 100 °C ([TMC]0/[l-LA]0/[PD]0/[CH3COONa] = 50/50/1/0.5) to extend the PLLA chains from the preformed PTMC-diol. After 24 h of
reaction, the monomer conversion of l-LA reached 66.6%, yielding an elastic material with a narrow _Ð_ value of 1.12. It should be noted that the unreacted TMC was not consumed during the
2nd polymerization step. Importantly, the SEC elution peak of the final product was observed in a higher molecular weight region than that of the preformed PTMC-diol, confirming the success
of the block copolymerization (Fig. 4(b)). In addition, the signal arising from the methylene groups adjacent to the _ω_-chain end hydroxyl groups in the PTMC-diol (3.74 ppm) was not
detected in the 1H NMR spectrum of the final product (Fig. 4(c)). Furthermore, new signals attributable to the TMC–l-LA linkages were observed at 5.10 (_h_), 4.45 (_g_), and 4.35 ppm (_e_).
Importantly, the integration ratio of the linkage signal (e.g., proton _h_) to the PLLA methine signal (_j_) agreed with the estimated value, which excluded the possibility of
transesterification between the PTMC and PLLA segments. These results indicated that the ROP of l-LA was initiated by the terminal hydroxyl groups of the PTMC-diol to afford the desired
PLLA-_b_-PTMC-_b_-PLLA triblock copolymer. The corresponding 13C NMR spectrum was reasonably assigned to the expected structure, though the signals attributable to the TMC–l-LA linkages were
not detected due to its small fraction. These results suggested the usefulness of the developed ROP system for the synthesis of functionalized APC materials for a wide range of
applications. CONCLUSIONS Herein, we established a highly efficient solvent-free ROP system for TMC using alkali metal carboxylate catalysts, which satisfies the industrial requirements of
low cost, easy handling, and safety. Importantly, the alkali metal carboxylates promoted the ROP of TMC even at low catalyst loadings of 0.001–0.0001 mol%, achieving the optimum TOF value
(34 200 h−1) using CH3COONa as the catalyst. This ROP system provides well-defined PTMCs with a predictable molecular weight and narrow dispersity. Furthermore, we successfully synthesized
end-functionalized PTMCs and PTMC-containing triblock copolymer using the CH3COONa catalyst with functional alcohol initiators. Given the solvent-free process and low catalyst loading, the
developed ROP system is amenable to the practical synthesis of APC-based materials for use in biomedical and environmental applications. One of the advantages of using alkali metal
carboxylates is the ability to fine-tune the catalytic performance by selecting the carboxylate moiety and countercation [19]. Further optimization of the catalyst would enable suppression
of the side reactions, including intermolecular transesterification. REFERENCES * Ulery BD, Nair LS, Laurencin CT. Biomedical applications of biodegradable polymers. J Polym Sci, Part B
Polym Phys. 2011;49:832–64. Article CAS Google Scholar * Taraghi I, Paszkiewicz S, Grebowicz J, Fereidoon A, Roslaniec Z. Nanocomposites of polymeric biomaterials containing carbonate
groups: an overview. Macromol Mater Eng. 2017;302:1–22. Article Google Scholar * Nair LS, Laurencin CT. Biodegradable polymers as biomaterials. Prog Polym Sci. 2007;32:762–98. Article CAS
Google Scholar * Brannigan RP, Dove AP. Synthesis, properties and biomedical applications of hydrolytically degradable materials based on aliphatic polyesters and polycarbonates. Biomater
Sci. 2017;5:9–21. Article CAS Google Scholar * Walter E, Moelling K, Pavlovic J, Merkle HP. Microencapsulation of DNA using poly(dl -lactide-co-glycolide): stability issues and release
characteristics. J Control Release. 1999;61:361–74. Article CAS Google Scholar * Tinsley-Bown AM, Fretwell R, Dowsett AB, Davis SL, Farrar GH. Formulation of poly(d,l-lactic-co-glycolic
acid) microparticles for rapid plasmid DNA delivery. J Control Release. 2000;66:229–41. Article CAS Google Scholar * Zhang Z, Kuijer R, Bulstra SK, Grijpma DW, Feijen J. The in vivo and
in vitro degradation behavior of poly(trimethylene carbonate). Biomaterials. 2006;27:1741–8. Article CAS Google Scholar * Ma Z, Hong Y, Nelson DM, Pichamuthu JE, Leeson CE, Wagner WR.
Biodegradable polyurethane ureas with variable polyester or polycarbonate soft segments: effects of crystallinity, molecular weight, and composition on mechanical properties.
Biomacromolecules. 2011;12:3265–74. Article CAS Google Scholar * Kricheldorf HR, Lossin M, Mahler A. Ring-opening polymerization of cyclobis (diethy1ene glycol carbonate) by means of
BuSnC13, SnOct2 or Bu2SnO as catalysts. Macromol Chem Phys. 1997;3570:3559–70. Article Google Scholar * Langlais M, Coutelier O, Moins S, Winter JD, Coulembier O, Destarac M. Scope and
limitations of ring-opening copolymerization of trimethylene carbonate with substituted γ-thiolactones. Polym Chem. 2018;9:2769–74. Article CAS Google Scholar * Kricheldorf HR, Stricker
A. Polymers of carbonic acid 29. Bu2SnOct2-initiated polymerizations of trimethylene carbonate (TMC, 1,3-dioxanone-2). Polym (Guildf). 2000;41:7311–20. Article CAS Google Scholar * Wurm
B, Keul H, Höcker H. Polymerization of 2,2‐dimethyltrimethylene carbonate with tri‐_sec_‐butoxy aluminium; a kinetic study. Macromol Chem Phys. 1994;195:3489–98. Article CAS Google Scholar
* Nederberg F, Lohmeijer BGG, Leibfarth F, Pratt RC, Choi J, Dove AP, et al. Organocatalytic ring-opening polymerization of trimethylene carbonate. Biomacromolecules. 2007;8:153–60.
Article CAS Google Scholar * Delcroix D, Martín-Vaca B, Bourissou D, Navarro C. Ring-opening polymerization of trimethylene carbonate catalyzed by methanesulfonic acid: activated monomer
versus active chain end mechanisms. Macromolecules. 2010;43:8828–35. Article CAS Google Scholar * Wang X, Cui S, Li Z, Kan S, Zhang Q, Zhao C, et al. A base-conjugate-acid pair for
living/controlled ring-opening polymerization of trimethylene carbonate through hydrogen-bonding bifunctional synergistic catalysis. Polym Chem. 2014;5:6051–9. Article CAS Google Scholar
* Makiguchi K, Ogasawara Y, Kikuchi S, Satoh T, Kakuchi T. Diphenyl phosphate as an efficient acidic organocatalyst for controlled/living ring-opening polymerization of trimethylene
carbonates leading to block, end-functionalized, and macrocyclic polycarbonates. Macromolecules. 2013;46:1772–82. Article CAS Google Scholar * Saito T, Takojima K, Oyama T, Hatanaka S,
Konno T, Yamamoto T, et al. Trimethyl glycine as an environmentally benign and biocompatible organocatalyst for ring-opening polymerization of cyclic carbonate. ACS Sustain Chem Eng.
2019;7:8868–75. Article CAS Google Scholar * Saito T, Aizawa Y, Tajima K, Isono T, Satoh T. Organophosphate-catalyzed bulk ring-opening polymerization as an environmentally benign route
leading to block copolyesters, end-functionalized polyesters, and polyester-based polyurethane. Polym Chem. 2015;6:4374–84. Article CAS Google Scholar * Saito T, Aizawa Y, Yamamoto T,
Tajima K, Isono T, Satoh T. Alkali metal carboxylate as an efficient and simple catalyst for ring-opening polymerization of cyclic esters. Macromolecules. 2018;51:689–96. Article CAS
Google Scholar * Shibasaki Y, Sanda F, Endo T. Activated monomer cationic polymerization of 1,3-dioxepan-2-one initiated by water-hydrogen chloride. Macromol Rapid Commun. 1999;20:532–5.
Article CAS Google Scholar * Kim J-H, Lee SY, Chung DJ. Synthesis and properties of triblock copolymers from L-Lactide and trimethylene carbonate. Polym J. 2002;32:1056–9. Article Google
Scholar * Palard I, Schappacher M, Belloncle B, Soum A, Guillaume SM. Unprecedented polymerization of trimethylene carbonate initiated by a samarium borohydride complex: mechanistic
insights and copolymerization with ε-caprolactone. Chem A Eur J. 2007;13:1511–21. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS This work was financially supported by
the JSPS KAKENHI (Grant Number JP18H04639) (Hybrid Catalysis for Enabling Molecular Synthesis on Demand), Frontier Chemistry Center (Hokkaido University), Inamori Foundation, and
Grant-in-Aid for JSPS Research Fellows. TS gratefully acknowledges the JSPS Fellowship for Young Scientists. VL, PB, and NH gratefully acknowledge the support of King Abdullah University of
Science and Technology. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Graduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo, 060-8628, Japan Kaoru Takojima &
Tatsuya Saito * Polytech Montpellier, 34095, Montpellier, France Cedric Vevert * Physical Sciences and Engineering Division, KAUST Catalysis Center, Polymer Synthesis Laboratory, King
Abdullah University of Science and Technology (KAUST), Thuwal, 23955, Saudi Arabia Viko Ladelta, Panayiotis Bilalis & Nikos Hadjichristidis * R&D Center, Organic Chemical Products
Company, Daicel Corporation, Hiroshima, 739-0695, Japan Jun Watanabe, Shintaro Hatanaka & Takashi Konno * Division of Applied Chemistry, Faculty of Engineering, Hokkaido University,
Sapporo, 060-8628, Japan Takuya Yamamoto, Kenji Tajima, Takuya Isono & Toshifumi Satoh Authors * Kaoru Takojima View author publications You can also search for this author inPubMed
Google Scholar * Tatsuya Saito View author publications You can also search for this author inPubMed Google Scholar * Cedric Vevert View author publications You can also search for this
author inPubMed Google Scholar * Viko Ladelta View author publications You can also search for this author inPubMed Google Scholar * Panayiotis Bilalis View author publications You can also
search for this author inPubMed Google Scholar * Jun Watanabe View author publications You can also search for this author inPubMed Google Scholar * Shintaro Hatanaka View author
publications You can also search for this author inPubMed Google Scholar * Takashi Konno View author publications You can also search for this author inPubMed Google Scholar * Takuya
Yamamoto View author publications You can also search for this author inPubMed Google Scholar * Kenji Tajima View author publications You can also search for this author inPubMed Google
Scholar * Nikos Hadjichristidis View author publications You can also search for this author inPubMed Google Scholar * Takuya Isono View author publications You can also search for this
author inPubMed Google Scholar * Toshifumi Satoh View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHORS Correspondence to Takuya Isono or
Toshifumi Satoh. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral
with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL MATERIAL RIGHTS AND PERMISSIONS Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Takojima, K., Saito, T., Vevert, C. _et al._ Facile synthesis of poly(trimethylene carbonate) by alkali metal carboxylate-catalyzed ring-opening
polymerization. _Polym J_ 52, 103–110 (2020). https://doi.org/10.1038/s41428-019-0264-6 Download citation * Received: 27 June 2019 * Revised: 18 August 2019 * Accepted: 24 August 2019 *
Published: 24 September 2019 * Issue Date: January 2020 * DOI: https://doi.org/10.1038/s41428-019-0264-6 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative