Targeting cell death pathways in intestinal ischemia-reperfusion injury: a comprehensive review
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Intestinal ischemia-reperfusion (I/R) is a multifaceted pathological process, and there is a lack of clear treatment for intestinal I/R injury. During intestinal I/R, oxidative
stress and inflammation triggered by cells can trigger a variety of cell death mechanisms, including apoptosis, autophagy, pyroptosis, ferroptosis, and necrosis. These cell death processes
can send a danger signal for the body to be damaged and prevent intestinal I/R injury. Therefore, identifying key regulatory molecules or markers of these cell death mechanisms when
intestinal I/R injury occurs may provide valuable information for the treatment of intestinal I/R injury. This paper reviews the regulatory molecules and potential markers that may be
involved in regulating cell death during intestinal I/R and elaborates on the cell death mechanism of intestinal I/R injury at the molecular level to provide a theoretical basis for
discovering new molecules or markers regulating cell death during intestinal I/R injury and provides ideas for drug development for the treatment of intestinal I/R injury. SIMILAR CONTENT
BEING VIEWED BY OTHERS MTDNA-STING PATHWAY PROMOTES NECROPTOSIS-DEPENDENT ENTEROCYTE INJURY IN INTESTINAL ISCHEMIA REPERFUSION Article Open access 11 December 2020 DIFFERENT TYPES OF CELL
DEATH AND THEIR INTERACTIONS IN MYOCARDIAL ISCHEMIA–REPERFUSION INJURY Article Open access 05 March 2025 THE EMERGING ROLE OF FERROPTOSIS IN INTESTINAL DISEASE Article Open access 17 March
2021 FACTS * Gaining a deeper understanding of the intricacies surrounding intestinal ischemia-reperfusion injury: elucidating the mechanisms of cell death. * Emerging therapeutic strategies
for mitigating intestinal ischemia-reperfusion injury. * To explore new therapeutic targets for intestinal ischemia-reperfusion injury from the perspective of oxidative stress and
inflammation. OPEN QUESTIONS * What is the etiology of intestinal ischemia-reperfusion injury and the efficacy of therapeutic interventions? * Which additional molecular processes
contributing to intestinal ischemia-reperfusion injury remain undisclosed? * Have reliable biomarkers been identified for intestinal ischemia-reperfusion injury, and are there still
challenges present in this particular domain of research? INTRODUCTION Intestinal ischemia-reperfusion (I/R) injury is a prevalent acute and critical condition in the clinical setting. It is
strongly associated with the onset, progression, and prognosis of various clinical illnesses [1,2,3]. Intestinal I/R injury is exogenous damage to the intestine that results in fluid
extravasation through capillaries and subsequent interstitial edema [4]. Following ischemia and subsequent reperfusion, the permeability of the intestinal capillary increases [5]. Mucosal
injury is characterized by the detachment of epithelial cells from the villi, necrosis of epithelial tissue, degradation of the lamina propria, hemorrhaging, and ulceration. These
pathological changes together lead to adverse consequences, such as a decrease in nutrient absorption and an increase in the permeability of the mucosal barrier, which facilitates the
passage of macromolecules [4, 6]. The activation of self-protective mechanisms in gut cells is a response to and a preventive measure against adverse outcomes [7, 8]. However, a definitive
program and standard for the clinical management of intestinal I/R injury are lacking. When intestinal I/R injury occurs, a cascade of cell death mechanisms is initiated in the body in
response to stress. The initiation of these cell death mechanisms is closely related to both the ischemia and reperfusion stages [9, 10]. Intestinal ischemia occurs due to reduced or
interrupted blood supply to the intestine, leading to insufficient oxygen and nutrient delivery to cells. This results in the disruption of energy metabolism and impairment of mitochondrial
function. Within this ischemic milieu, cellular metabolic activities are compromised, the energy supply is disrupted, and cellular damage occurs [4, 11, 12]. The activation of multiple
signaling pathways leads to the initiation of various mechanisms of cell death. Upon reperfusion of blood into the intestine, compromised cells are exposed to an oxygen-enriched milieu,
which promotes an inflammatory reaction facilitated by the accumulation of metabolites generated during ischemia and the release of oxygen free radicals [13]. These inflammatory responses
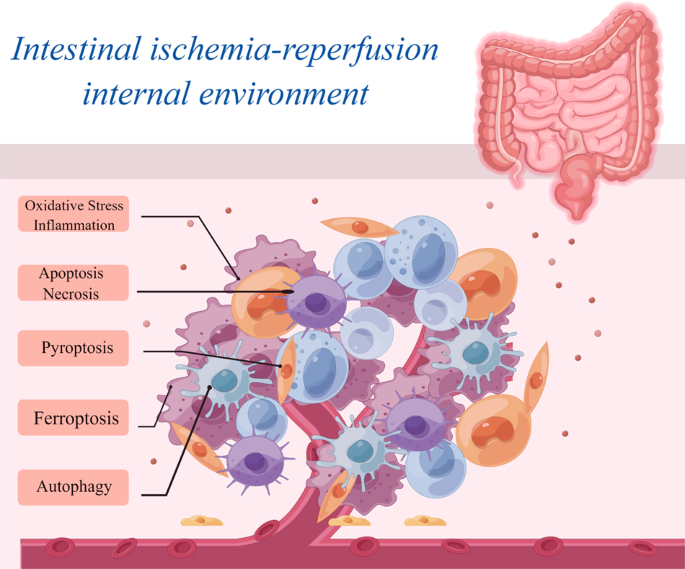
can induce cell death by activating factors and signaling pathways linked to different forms of cell death. The mechanisms underlying cell death related to intestinal I/R injury can be
categorized primarily into programmed and non-programmed forms. Programmed cell death includes apoptosis, autophagy, pyroptosis, and ferroptosis, whereas non-programmed cell death primarily
refers to necrosis. However, whether programmed cell death or non-programmed cell death occurs, these cell death pathways cannot operate autonomously in the context of intestinal I/R injury
[14, 15]. In individuals with intestinal ischemia, affected cells experience inadequate oxygen and nutrient supplies, leading to energy metabolism disturbances and an unstable intracellular
milieu. These changes promote programmed cell death [16]. During post-ischemic reperfusion, stimulation and damage to the intestine increase, primarily due to the buildup of metabolites
generated during ischemia and the release of oxygen free radicals. Under such conditions, the gut undergoes non-programmed cell death. Although a direct causal connection between these forms
of cell death has not been determined, they can also interact [17, 18]. Thus, the intricate interplay between different cell death modalities and their corresponding target genes in the
context of intestinal I/R injury needs to be elucidated and summarized. In this study, we comprehensively analyzed the classical cellular mechanisms associated with intestinal I/R that have
been described in recent years. We examined and integrated potential targets associated with these mechanisms. This study might provide a foundation for future research in the field of
intestinal I/R injury. OXIDATIVE STRESS AND INFLAMMATION Oxidative stress is characterized by an imbalance between oxidation and antioxidant processes in the body. The condition promotes
oxidation and the infiltration of neutrophils, an increase in the secretion of proteases, and the generation of numerous oxidative intermediates [19]. The body produces reactive oxygen
species (ROS) and reactive nitrogen species (RNS) as a response to detrimental stimuli originating from internal and external environments. [19, 20] Inflammation is a ubiquitous
physiological response that occurs in vascularized living tissues as a defensive mechanism to protect against various injurious stimuli. It occurs in almost all instances of tissue injury in
humans, including infections, trauma, diseases, and other stimuli that elicit a bodily reaction. Thus, it is a universally occurring phenomenon [21, 22]. Oxidative stress and inflammation,
which affect each other under physiological and pathological conditions and jointly participate in the occurrence and development of many diseases, are closely related. Oxidative stress can
cause damage to biomolecules such as cell membranes, DNA, and proteins, thus triggering an inflammatory response. During inflammation, white blood cells release a range of ROS and free
radicals to destroy pathogens or remove damaged tissue. However, an excessive inflammatory response can also lead to an increase in oxidative stress. Overall, maintaining the redox balance
in the body and a moderate inflammatory response are essential for health [23, 24]. INTESTINAL I/R AND OXIDATIVE STRESS During intestinal ischemia, an insufficient blood supply impedes
oxygen transport to intestinal cells, which causes a poor oxygen supply to cells and hinders normal oxidative metabolism. Consequently, mitochondria play a key role in REDOX reactions and
produce a large number of oxygen free radicals and other ROS under hypoxic conditions [25, 26]. When blood is reintroduced into ischemic intestinal tissue, it replenishes oxygen in the
cells. However, the surplus oxygen supplied during this period might induce oxidative stress [27, 28]. Hence, the induction of oxidative stress during intestinal I/R injury may trigger a
cascade of signaling pathways and inflammatory reactions, including nuclear factor-κB (NF-κB), NADPH oxidase (NOX), and apoptosis signal-regulated kinase (ASK). After activation, these
pathways exacerbate oxidative stress, which results in a detrimental cycle, eventually leading to cell impairment and death. INTESTINAL I/R AND INFLAMMATION Intestinal I/R injury-induced
inflammation is mainly related to the release of inflammatory mediators, the activation and infiltration of leukocytes, and the release of ROS [29,30,31]. Intestinal I/R induces the
synthesis and release of inflammatory mediators, such as cytokines and chemical mediators [32, 33]. These inflammatory mediators elicit vascular dilation, leukocyte infiltration, platelet
activation, and other inflammatory responses, which exacerbate the impairment of the intestinal mucosal barrier and promote the progression of inflammatory reactions [31, 34]. The presence
of inflammatory cells, inflammatory mediators, and free radicals can harm the structural integrity of intestinal tissue, which compromises the integrity of the intestinal mucosal barrier and
increases mucosal permeability [30, 35]. This process facilitates the infiltration of intestinal bacteria and toxins and subsequently leads to the activation of the immune system and
pro-inflammatory cells. These changes eventually lead to inflammatory damage [36, 37]. OXIDATIVE STRESS AND INFLAMMATION IN INTESTINAL I/R INJURY Hydrogen peroxide (H2O2)-induced
mitochondrial oxidative damage and apoptosis are major risk factors for intestinal I/R injury [38]. A study on the underlying mechanism showed that peroxiredoxin 3 (PRDX3) is involved in
intestinal I/R injury (Fig. 1). PRDX3 decreased sirtuin-3 (SIRT3) expression during infection and increased acetylation in Caco-2 cells. The inhibition of SIRTs by nicotinamide (NAM)
increased the level of acetylated PRDX3 and impaired its antioxidant activity. Additionally, SIRT3 deacetylation at lysine residue K253 increased in mice, which increased SIRT3 dimerization.
Transfection of the K253Q plasmid decreased after 12 h of hypoxia, and 4 h of reoxygenation [8]. Thus, oxidative stress regulates ROS production and intestinal I/R injury [39]. CircRNA
sponges can reduce hypoxia/reoxygenation (H/R)-induced ROS overproduction by decreasing mitochondrial superoxide anion (O2−) levels and NADPH oxidase activity and increasing the expression
of antioxidant enzymes (Fig. 1). In vitro, H/R was performed in Caco-2 cells after intravenous injection of miR-339-5p agomir or circ-protein kinase C beta (PRKCB) siRNA. The enhanced PRKCB
gene-transcribed circRNA circ-PRKCB acted as an endogenous miR-339-5p sponge to regulate the expression of p66Shc and reduce oxidative stress and I/R injury in vivo and in vitro following
intestinal infarction. The study’s findings indicated that circ-PRKCB/miR-339-5p/p66Shc is important for understanding oxidative stress related to intestinal I/R [40]. Several studies on
intestinal I/R injury have shown that certain monomers derived from traditional Chinese medicine or non-coding RNAs can regulate intestinal I/R injury via mechanisms involving oxidative
stress or the inflammatory response. For example, agmatine can mitigate intestinal I/R injury in rats by reducing oxidative stress and inflammatory responses [41]. Similarly, dioscin can
attenuate intestinal I/R injury in mice by modulating oxidative stress mediated by miR-351-5p [42] (Fig. 1). In contrast, miR-23a-5p can promote oxidative stress and exacerbate intestinal
I/R injury in mice by targeting peroxisome proliferator-activated receptor alpha (PPARα) [43] (Fig. 1). The activation of the nuclear factor erythroid 2-related factor 2 (Nrf2)/heme
oxygenase-1 (HO-1)/NAD(P)H: quinone acceptor oxidoreductase 1 (NQO1) signaling pathway regulates the anti-inflammatory, antioxidant, and anti-apoptotic effects induced by sesamin; thus, this
pathway protects against and alleviates intestinal I/R injury in rats [44] (Fig. 1). These findings indicate that oxidative stress and inflammation are involved in intestinal I/R injury.
These findings will also contribute to the identification of novel therapeutic targets for treating this pathological condition. PROGRAMMED AND NON-PROGRAMMED CELL DEATH Programmed cell
death primarily occurs during the growth and development of certain organisms. Based on certain predetermined requirements, some cells are sacrificed to meet a specific goal [45]. In
contrast, in non-programmed cell death, an organism is compelled to sacrifice specific cells due to external factors or certain reasons, often resulting from physiological irregularities
[46, 47]. The distinguishing features of programmed and non-programmed cell death are summarized in Fig. 2. When intestinal I/R injury occurs, a large number of impaired cells undergo
apoptosis [48, 49], and the reliance on a single mode of cell death is a serious threat to the intestinal tissue, as inhibiting this pathway can lead to grave consequences. Preserving
intestinal integrity requires the action of programmed and non-programmed cell death mechanisms. APOPTOSIS AND NECROSIS Apoptosis is a regulated form of cell death, which mainly occurs
through the activation of the Caspase protease family, leading to the fragmentation of nuclear DNA and eventually cell death. Specifically, apoptosis activates the Caspase protease family
through two main pathways (endogenous and exogenous). In the endogenous pathway, mitochondria release cytochrome C and activate Caspase 9. In an exogenous pathway, the death receptor
activates Caspase 8. The activated initiating Caspase eventually activates the executive Caspase, resulting in nuclear DNA breakage and ultimately apoptosis [50]. This process plays a key
role in maintaining intracellular homeostasis and facilitates intricate operations in multicellular organisms [51]. Apoptosis is an important process that occurs in intestinal tissue. It
helps eliminate non-functional, unwanted, abnormal, and detrimental cells. By optimizing the intestinal structure and cell count, apoptosis ensures the appropriate development of the
intestine [52, 53]. Necrosis is an uncontrolled, passive process of cell death. It is usually caused by severe cell damage, a process in which cells are irreversibly damaged and eventually
die during a pathological process. Necrosis is characterized by swelling of organelles, damage to the plasma membrane, and eventually, cell lysis. Cell contents spill into the surrounding
tissue, causing damage to the tissue. Unlike programmed cell death, cell necrosis is primarily caused by harmful agents that invade the body [54, 55]. RELATIONSHIP OF INTESTINAL I/R WITH
APOPTOSIS AND NECROSIS Ischemia can stimulate the production of deleterious substances and also induce inflammation, apoptosis, and necrosis of epithelial cells [56]. Intestinal I/R injury
induces apoptosis and necrosis in the intestinal epithelial cells, which results in impaired intestinal barrier function and ultimately leads to multiple organ dysfunction syndrome [49]. The
inflammatory response drives the occurrence of apoptosis and necrosis [57]. Inflammatory mediators and cytokines released during intestinal I/R injury can trigger the activation of
signaling pathways associated with apoptosis and necrosis, thereby facilitating the onset of apoptosis and necrosis [58]. The target genes or signaling pathways that can regulate apoptosis
and necrosis in intestinal I/R injury include a prolyl-isomerase, peptidyl-prolyl cis-trans isomerase (Pin1), and p66Shc (pink arrow; Fig. 3), SIRT3 and PRDX (the brown arrow, Fig. 3), the
SIRT3 and Putative kinase 1 (PINK1)/Histone deacetylase 3 (HDAC3)/p53 (blue arrow; Fig. 3) signaling pathway, the miR-351–5p/mitogen-activated protein kinase (MAPK13) (purple arrow; Fig. 3)
signaling pathway, the miR-29b-3p/TNF receptor-associated factor 3 (TRAF3)/TGF-α-activated kinase 1 (TAK1) (red arrow; Fig. 3) signaling pathway, etc. Some drugs, such as Dexmedetomidine and
dioscin, can also regulate apoptosis and necrosis associated with intestinal I/R injury. APOPTOSIS AND NECROSIS IN INTESTINAL I/R INJURY Intestinal epithelial oxidative stress and apoptosis
are the key pathogenic mechanisms underlying intestinal I/R injury. Pin1 was found to regulate the activity of p66Shc during intestinal I/R. This upregulation led to the accumulation of
intestinal mitochondrial ROS and the apoptosis of many epithelial cells. Additionally, it increased protein expression and enzyme activity of Pin1, as well as, the interaction between Pin1
and p66Shc. The activation of Pin1 facilitated the translocation of p66Shc to the mitochondria during intestinal I/R, and it also helped to alleviate gut damage and secondary lung injury.
These findings suggested that Pin1 inhibition may be a novel prophylactic target for treating intestinal I/R injury [38]. Some traditional Chinese medicine monomers or non-coding RNAs can
regulate intestinal I/R injury through apoptosis or necrosis. For example, Dexmedetomidine can protect rats against intestinal I/R injury by promoting mitophagy and inhibiting the apoptosis
of enteric glial cells (EGCs) through inhibit the SIRT3/PINK1/HDAC3/p53 pathway [59]. Dioscin can alleviate intestinal I/R injury in mice by regulating miR-351-5p/MAPK13-mediated
inflammation and apoptosis [60]. SIRT3-mediated deacetylation of PRDX3 can decrease mitochondrial oxidative damage and apoptosis induced by intestinal I/R injury in mice [8]. The findings of
these studies indicated that apoptosis and necrosis can significantly contribute to intestinal I/R injury. In conclusion, studies on intestinal I/R injury and its association with apoptosis
and necrosis have found that during the ischemic phase, cellular damage occurs, followed by inflammatory responses during reperfusion, which leads to tissue necrosis [58, 61]. Inflammation
plays a key role in intestinal I/R injury, regardless of whether apoptosis or necrosis is involved. To better understand the mechanisms underlying apoptosis and necrosis in intestinal I/R
injury, changes in inflammatory markers need to be monitored. The association between intestinal I/R injury and cell death mechanisms like apoptosis and necrosis needs to be further studied.
PYROPTOSIS Pyroptosis is a mode of programmed death of inflammatory cells that occurs mainly through the activation of various Caspases, including Caspase 1, mediated by inflammatory
bodies. Pyroptosis leads to the shear and polyaggregation of various Gasdermin family members, including gasdermin D (GSDMD), resulting in cell perforation and cell death [62]. Pyroptosis is
faster than apoptosis and is accompanied by the release of a large number of pro-inflammatory cytokines [63]. When pyroptosis occurs, cells swell. Before the cell ruptures, protrusions form
on the cell, and pores form on the cell membrane, which causes the cell membrane to lose its integrity and release its contents, resulting in inflammation. Under such conditions, the
nucleus is located in the center of the cell [62, 64]. INTESTINAL I/R AND PYROPTOSIS In intestinal I/R, hypoxia, and reperfusion cell damage may cause pyroptosis. Unlike apoptosis, which is
a controlled cell death process, pyroptosis is an uncontrolled form of cell death that occurs when the gut is severely damaged by cells. Pyroptosis is a major risk factor for intestinal
barrier destruction and cell death [65]. The occurrence of pyroptosis in intestinal I/R injury was confirmed by histopathological findings and intestinal barrier indices, including
transepithelial electrical resistance (TER), tight-junction protein, and serum biomarkers. These findings indicated that I/R injury caused intestinal barrier destruction and cell death [65].
Studies on pyroptosis have focused on the classical signaling pathway induced by exogenous injury [62], although both classical and non-classical signaling pathways are present.
Understanding the link between intestinal I/R and pyroptosis can help in developing preventive and therapeutic strategies to reduce the adverse effects of intestinal I/R injury in patients.
Recent studies on intestinal I/R injury have identified several target genes and signaling pathways involved in the regulation of pyroptosis, including Thioredoxin-interacting protein
(TXNIP) (light blue arrow; Fig. 4), miR-122a, and epidermal growth factor receptor (EGFR) (purple arrow; Fig. 4), Toll-like receptor4 (TLR4), Toll/IL-1 receptor domain-containing adapter
(TRIF), receptor-interacting serine/threonine-protein kinase-1 (RIPK1), RIPK3 (dark green arrow; Fig. 4), ROS (blue arrow; Fig. 4), etc. The traditional Chinese medicine Corilagin (Cor) can
also treat intestinal I/R injury by regulating pyroptosis. PYROPTOSIS IN INTESTINAL I/R INJURY The hypoglycemic drug metformin protects the barrier function of intestinal I/R injury by
controlling pyroptosis [65]. Additionally, it actively participates in the suppression of pyroptosis-associated proteins, such as NLRP3 (NOD-, LRR- and pyrin domain-containing 3), Cleaved
Caspase 1, and the N-terminus of GSDMD. Metformin can suppress the expression of TXNIP and its interaction with NLRP3. The protective effects of metformin disappeared by siRNA knockdown.
This implies that the primary mechanism by which metformin exerts its protective effects against intestinal I/R injury is through its interaction with TXNIP [65]. This was the first study in
the field of intestinal I/R injury to show that pyroptosis is involved in the process of injury. This finding provided the foundation for subsequent research on the role of pyroptosis in
intestinal I/R injury. A study found that increasing the expression of miR-122a significantly inhibits the activity of EGFR in intestinal I/R injury, reduces the expression of EGFR mRNA and
protein, and increases the expression of NLRP3 mRNA and protein. The expression of Caspase 1, N-GSDMD, ASC, IL-1β, and IL-18 proteins was upregulated to promote pyroptosis [66]. Thus,
miR-122a is essential for regulating intestinal I/R injury; the study also found that miR-122a promotes pyroptosis by inhibiting the EGFR-NLRP3 signaling pathway in mice [66]. Cor is a
natural ellagitannin found in various plants and has many biological and pharmacological properties [67]. It can significantly reduce intestinal I/R-induced pathological injury, inflammatory
response, oxidative stress, NLRP3 inflammasome activation, and pyroptosis in intestinal and lung tissues both in vivo and in vitro. Thus, Cor may be an effective therapeutic agent for
treating intestinal I/R-induced inflammation and tissue injury [68]. These studies focused on investigating the mechanism by which pyroptosis affects intestinal I/R injury and identifying
potential therapeutic targets. The findings of the above-mentioned studies indicated that the assembly and activation of the NLRP3 inflammasome strongly influence the initiation of
pyroptosis, irrespective of alterations in the target genes or pathways governing pyroptosis [69, 70]. Hence, alterations in the NLRP3 inflammasome need to be elucidated to identify
potential targets for the regulation of pyroptosis. Although the investigation of the effects of pyroptosis on intestinal I/R injury started later than studies on apoptosis and necrosis, it
has received increasing attention. Pyroptosis is considered to be an important mechanism in the study of intestinal I/R injury. FERROPTOSIS Ferroptosis is a regulated form of cell death
characterized by a lethal threshold of iron-dependent increase in lipid peroxidation. The inhibition of cystine transport proteins, such as Erastin, leads to the depletion of intracellular
Glutathione (GSH), which leads to the inactivation of Glutathione Peroxidase 4 (GPX4) [71, 72]. This inactivation increases lipid peroxidation, which partially induces cell death. The direct
inhibition of GPX4, caused by RSL3, also leads to this effect [73, 74]. Several biological processes are highly susceptibility to ferroptosis, including the metabolism of amino acids, iron,
and polyunsaturated fatty acids, as well as the biosynthesis of GSH, phospholipids, NADPH, and coenzyme Q10 [73, 75]. Several studies have found a correlation between intestinal I/R injury
and ferroptosis, and some studies have also provided evidence that ferroptosis is involved in intestinal I/R injury [74, 76]. INTESTINAL I/R AND FERROPTOSIS The main factors contributing to
ferroptosis in intestinal I/R injury include oxidative stress, the release of iron ions, lipid peroxidation, and iron dependence [76,77,78,79]. Intestinal I/R injury induces oxidative
stress, characterized by the excessive accumulation of reactive oxygen species generated intracellularly. Excessive ROS production disrupts the cellular REDOX equilibrium [79]. Iron, which
serves as a key catalyst and oxidizing agent, participates in the generation of free radicals, thereby increasing oxidative stress [80]. Iron ions are released after cell injury and the
subsequent disruption of cell membranes and histiocytosis during intestinal I/R [78]. Iron ions, in an unbound state, can interact with ROS and enhance oxidative stress. Iron-induced
oxidative stress increases lipid peroxidation [76, 81]. Under oxidative stress, a large number of ROS can engage in reactions that disrupt the integrity of cellular lipid structures, which
damage and impair the functions of the cell membrane [26]. The occurrence of ferroptosis depends on the presence of free iron in the cellular milieu. In the context of intestinal I/R injury,
ferroptosis might be a prominent mechanism of cell death caused by the disruption of tissue integrity and the subsequent release of free iron ions [76]. Studies have identified specific
genes and signaling pathways, such as special protein 1 (Sp1) (pink arrow; Fig. 5), transient receptor potential cation channel subfamily V member 1 (TRPV1) (brown arrow; Fig. 5), the Nrf2
signaling pathway (dark blue arrow; Fig. 5), HO-1 (green arrow; Fig. 5), the Nrf2/telomerase reverse transcriptase (TERT) signaling pathway (purple arrow, Fig. 5), and monoamine oxidase b
(MAO-B) (blue arrow; Fig. 5), which can regulate ferroptosis associated with intestinal I/R injury. Additionally, certain traditional Chinese medicines, such as capsiate (CAT) and
Apigenin-7-O-β-D-(-6″-p-coumaroyl)-glucopyranoside (APG), can regulate ferroptosis induced by intestinal I/R injury. FERROPTOSIS IN INTESTINAL I/R INJURY Acyl-CoA synthetase long-chain
family member 4 (ACSL4) is a key enzyme that regulates lipid composition during reperfusion. Sp1 is a crucial transcription factor that can increase the transcription of ACSL4 by binding to
the ACSL4 promoter region. This interaction provides a unique and effective mechanism for preventing intestinal I/R injury by controlling ferroptosis [76]. Some intestinal metabolites or
traditional Chinese medicine monomers can also regulate intestinal I/R injury through ferroptosis. For example, some studies investigated the changes in intestinal flora and metabolites in
the process of intestinal I/R [3, 82, 83], as well as, the protective effects of CAT. CAT is a natural plant compound related to chili peppers and belongs to the capsaicin family [84]. The
results showed that CAT is a metabolite of the intestinal flora, and preoperative fecal CAT level in patients with cardiopulmonary bypass is negatively correlated with intestinal I/R injury.
However, the glutathione peroxidase 4 (Gpx4) inhibitor RSL3 is inhibited by CAT. The TRPV1 antagonist JNJ-17203212 can enhance Gpx4 expression and inhibit iron poisoning by activating
TRPV1, which provides a potential pathway for intestinal I/R treatment [84]. APG is a new flavonoid glycoside isolated from _Clematis tangutica_ that has strong antioxidant abilities [85].
Studies have investigated the effect of APG on intestinal I/R in vivo and in vitro and found that APG can significantly improve intestinal edema and Chiu’s score in mice. It can reduce ROS
production and Fe2+ accumulation, maintain mitochondrial function, and inhibit ferroptosis [86]. These studies showed that ferroptosis is associated with intestinal I/R injury. Their
findings provide a fundamental basis for understanding the role of ferroptosis in this specific pathological condition. They also provide valuable insights into and avenues for future
investigations on the involvement of ferroptosis in intestinal I/R injury. Ferroptosis is a form of iron-dependent cell death [87]. To summarize, intestinal I/R-induced oxidative stress,
release of iron ions, lipid peroxidation, dependence on iron, and other factors may contribute to iron death [76, 88, 89]. Recent studies have shown that oxidative stress might be the most
plausible determinant of iron death in the context of intestinal I/R injury [79]. When investigating ferroptosis, the effect of oxidative stress needs to be accounted for, and pertinent
markers of oxidative stress need to be evaluated. However, ferroptosis, which is a novel form of cell death, has received limited attention in the context of I/R injury. Thus, its role and
underlying mechanisms associated with I/R injury need to be comprehensively investigated. AUTOPHAGY Autophagy is a vital intracellular degradation process through which damaged or excessive
proteins, organelles, and intracellular waste are sequestered within autophagosomes. These autophagosomes subsequently fuse with lysosomes, which leads to the breakdown of their contents
into essential molecular components that can be used for cellular recycling [90]. Autophagy helps maintain cellular integrity, facilitate cell viability during nutrient scarcity, and react
to cytotoxic agents. Autophagy includes constitutive autophagy, which operates under regular physiological circumstances, and inducible autophagy, which is activated in response to stress
[91, 92]. Constitutive autophagy helps in self-preservation by facilitating the growth and development of cells, protecting cells from metabolic stress and oxidative harm, and playing a
vital role in maintaining cellular homeostasis. It also helps in the synthesis, degradation, and recycling of cellular constituents. However, excessive autophagy can result in metabolic
stress, the degradation of cellular constituents and, ultimately, cell death [90, 92]. Several studies have shown that autophagy participates in various physiological and pathological
processes, including cellular equilibrium, aging, immune responses, tumor formation, and intestinal I/R injury [93, 94]. INTESTINAL I/R AND AUTOPHAGY Autophagy may be induced during
intestinal I/R to eliminate impaired cellular constituents and maintain cell viability [95]. Intestinal ischemia disrupts energy metabolism and destabilizes the intracellular milieu. Cells
may perform autophagy to eliminate dysfunctional or anomalous organelles and proteins, thus obtaining energy and nutrients through the degradation of these cellular components to ensure
survival. Upon reperfusion of blood flow into the ischemic intestine, an inflammatory response and cellular stress might occur, which might trigger autophagy as a cellular stress response
mechanism to regulate the intracellular environment and facilitate cellular adaptation [95, 96]. While examining intestinal I/R injury, autophagy flux was found to be impaired throughout the
intestinal I/R procedure [97]. Mitochondrial autophagy plays a key role in the pathogenesis of intestinal I/R injury [98, 99]. Some studies have provided evidence for the existence of
target genes or signaling pathways, such as the miR-665–3p/ATG4B (autophagy-related cysteine protein) signaling pathway (purple arrow; Fig. 6), the NOD-like receptor X1 (NLRX1)/FUN14
domain-containing 1 (FUNDC1)/nitrophenylphosphatase domain and non-neuronal SNAP25-like protein homolog 1 and 2 (NIPSNAP1/2) signaling pathway (green arrow; Fig. 6), the
miR-146a-5p/TXNIP/protein kinase AMP-activated catalytic subunit alpha (PRKAA)/mechanistic target of rapamycin kinase (mTOR) signaling pathway (brown arrow; Fig. 6), and the Glycogen
synthase kinase 3 beta (GSK-3β)/Nrf2 signaling pathway (blue arrow; Fig. 6). These pathways can regulate autophagy associated with intestinal I/R injury. Additionally, certain traditional
Chinese medicines, including paeoniflorin, can modulate autophagy during intestinal I/R injury [95]. AUTOPHAGY IN INTESTINAL I/R INJURY Autophagy is predominantly used along with traditional
Chinese medicine monomers or non-coding RNA for regulating diseases in studies on intestinal I/R injury. Paeoniflorin, a monoterpene glucoside, has various health benefits, including
regulation of autophagy and anti-inflammatory, anti-apoptotic, and antioxidant effects [100, 101]. Paeoniflorin preconditioning can protect rats against intestinal I/R injury by alleviating
intestinal histological damage, inflammatory response, oxidative stress, and cell apoptosis, without affecting control cells. Impairments in autophagy can be restored by upregulating the
degradation of autophagy-related proteins p62/SQSTM1, the expression of LC3-II and beclin-1, and the synthesis of autophagosome. Some [95] researchers have shown that miRNAs negatively
regulate autophagy by targeting ATGs [102]. Autophagy flux can be compromised during intestinal I/R. Additionally, miR-665–3p can negatively affect the expression of ATG4B in Caco-2 and
IEC-6 cells, which was shown using ileum biopsy samples obtained from patients diagnosed with intestinal infarction. In vivo, locked nucleic acid-modified inhibition reduces I/R-induced
systemic inflammation and apoptosis by restoring the autophagy flux [97]. In the mitochondria, NLRX1 is highly expressed in the gut and can regulate ROS production, mitochondrial damage,
autophagy, and apoptosis [103, 104]. Some studies examined the role of NLRX1 in mitochondrial homeostasis and apoptosis after intestinal I/R injury and found that NLRX1 was significantly
downregulated after intestinal I/R injury [98]. The miR-146a-5p/TXNIP axis can also reduce intestinal I/R injury in mice by inhibiting autophagy through the PRKAA/mTOR signaling pathway
[94]. Ischemic postconditioning (IPO) is a treatment method for I/R injury. The core of IPO is to mitigate reperfusion injury through short, alternating blood flow blockade and restoration
during the reperfusion stage [105]. Research has shown that IPO can effectively protect intestinal I/R injury from ischemic postconditioning by inducing autophagy, activating Akt,
inactivating GSK-3β, and activating Nrf2. These studies found that autophagy strongly influences the development and progression of intestinal I/R injury, serving as a well-established
cellular protective mechanism. To summarize, stimulating autophagy during intestinal I/R allows cells to cope with oxidative stress and other forms of damage, which reduces the likelihood of
cell death [65, 95]. The activation of autophagy in response to intestinal I/R serves as a cell survival mechanism. It facilitates the elimination of impaired cellular constituents and
cellular adaptation [65, 106]. A comprehensive understanding of the association between intestinal I/R and autophagy provides insights into the underlying pathophysiological mechanisms and
novel strategies for designing therapeutic interventions. DISCUSSION Intestinal I/R is the restoration of blood flow to the intestine following a period of inadequate blood supply, which can
occur in various clinical scenarios, such as intestinal obstruction, thrombosis, and insufficient arterial blood supply. The resultant tissue hypoxia and nutrient insufficiency stemming
from intestinal ischemia can lead to several pathophysiological mechanisms, including disturbances in cellular energy metabolism, impairment of mitochondrial function, and generation of
oxygen free radicals, among others [2, 77]. These alterations increase intracellular stress, subsequently resulting in cell death. Upon reperfusion, the ischemic region is replenished with
blood, which allows cells to regain access to oxygen and nutrients. However, this process might be harmful as the reintroduction of blood to ischemic tissue during reperfusion may lead to
reperfusion injury, and eventually, cell death [4, 107]. Several studies have shown that intestinal I/R injury induces cell death via two primary pathways, including oxidative stress and
inflammatory response [65, 107,108,109,110,111]. Upon reperfusion, oxygenated environments supply tissues with high levels of oxygen, which promotes the generation of oxygen free radicals.
These radicals, characterized by their high reactivity, engage with intracellular biological macromolecules, including proteins, lipids, and nucleic acids, thus increasing oxidation
reactions that lead to cell impairment and death. Oxidative stress negatively affects various cellular components, such as membrane lipids, mitochondria, and DNA, thus compromising their
structural integrity and hindering their functions. The cumulative effect of these direct and indirect consequences leads to cell death [65, 107, 110, 111]. Reperfusion following ischemia
can elicit an inflammatory response. After the reintroduction of blood flow to the ischemic region, immune cells, i.e., neutrophils and macrophages, become activated and release inflammatory
mediators, which include inflammatory cytokines and chemokines. The excessive release of these inflammatory mediators increases inflammatory reactions, which promotes the infiltration of
inflammatory cells and elicits a tissue inflammatory response, eventually leading to cell death [108, 109]. Intestinal I/R injury is closely associated with various modes of cell death,
where oxidative stress and inflammation serve as the underlying mechanisms following abnormal changes in the intestinal environment [40, 49, 109, 111, 112]. Several studies have found a
strong correlation between intestinal I/R and programmed cell death, specifically apoptosis. Intestinal ischemia disrupts the oxygen and nutrient supply to tissues, which compromises cell
survival and results in pathophysiological alterations, including energy metabolism disruption, mitochondrial dysfunction, and an increase in intracellular stress. These factors collectively
activate the apoptosis signaling pathway, which induces cells to enter the apoptotic state. When blood flows back into the ischemic intestinal tissue during reperfusion, the apoptotic
signaling pathway might be further activated, thus accelerating apoptosis. During reperfusion, oxidative stress and inflammation can influence apoptotic signaling pathways in various ways,
stimulating cells to enter an apoptotic state [49, 109, 112]. Several studies have found a correlation between intestinal I/R and non-programmed cell death. [113, 114] In contrast to
programmed cell death, such as apoptosis, non-programmed cell death does not follow a pre-established intracellular signaling cascade. Necrosis is a common form of non-programmed cell death.
During intestinal I/R, cells might get damaged due to ischemia, which can lead to non-programmed cell death during the subsequent reperfusion phase. Reperfusion injury manifests as
necrosis, characterized by non-programmed cell death. The incidence of reperfusion injury is closely associated with various factors, such as oxidative stress, inflammation, and
mitochondrial dysfunction. The detrimental effects of oxidative stress-induced oxygen free radicals and inflammatory response-released inflammatory mediators affect the cell membrane,
organelles, and key cellular molecules, which lead to non-programmed cell death [40, 111, 112]. To summarize, the occurrence of intestinal I/R can elicit cell death via oxidative stress and
inflammatory response. Oxygen free radicals induce oxidative stress, and along with inflammatory responses elicited by inflammatory mediators, they exert direct and indirect harmful effects
on cell structure and functionality, ultimately leading to cell death. Although we tried to clarify the mechanism of cell death induced by oxidative stress and inflammation during intestinal
I/R injury, we did not thoroughly analyze the specific induction, mechanism or potential target of cell death during intestinal I/R injury, and the cause of cell death has not been fully
elucidated. In future studies, more comprehensive studies on the triggers, mechanisms and targets of cell death during intestinal I/R generation are needed, as these studies may help
elucidate the complex mechanisms of intestinal I/R injury. Also, more targets need to be identified, and better therapeutic interventions need to be developed to address the unanswered
questions in this field of research. REFERENCES * Rodriguez-Lara SQ, Cardona-Munoz EG, Ramirez-Lizardo EJ, Totsuka-Sutto SE, Castillo-Romero A, Garcia-Cobian TA, et al. Alternative
interventions to prevent oxidative damage following ischemia/reperfusion. Oxid Med Cell Longev. 2016;2016:7190943. Article PubMed PubMed Central Google Scholar * Kalogeris T, Baines CP,
Krenz M, Korthuis RJ. Ischemia/reperfusion. Compr Physiol. 2016;7:113–70. Article PubMed PubMed Central Google Scholar * Deng F, Lin ZB, Sun QS, Min Y, Zhang Y, Chen Y, et al. The role
of intestinal microbiota and its metabolites in intestinal and extraintestinal organ injury induced by intestinal ischemia reperfusion injury. Int J Biol Sci. 2022;18:3981–92. Article CAS
PubMed PubMed Central Google Scholar * Hu J, Deng F, Zhao B, Lin Z, Sun Q, Yang X, et al. Lactobacillus murinus alleviate intestinal ischemia/reperfusion injury through promoting the
release of interleukin-10 from M2 macrophages via Toll-like receptor 2 signaling. Microbiome. 2022;10:38. Article CAS PubMed PubMed Central Google Scholar * Zhong J, Sun Y, Han Y, Chen
X, Li H, Ma Y, et al. Hydrogen sulfide-loaded microbubbles combined with ultrasound mediate thrombolysis and simultaneously mitigate ischemia-reperfusion injury in a rat hindlimb model. J
Thromb Haemost. 2021;19:738–52. Article CAS PubMed PubMed Central Google Scholar * Belle NM, Ji Y, Herbine K, Wei Y, Park J, Zullo K, et al. TFF3 interacts with LINGO2 to regulate EGFR
activation for protection against colitis and gastrointestinal helminths. Nat Commun. 2019;10:4408. Article ADS PubMed PubMed Central Google Scholar * Xiao L, Wu J, Wang JY, Chung HK,
Kalakonda S, Rao JN, et al. Long noncoding RNA uc.173 promotes renewal of the intestinal mucosa by inducing degradation of microRNA 195. Gastroenterology. 2018;154:599–611. Article CAS
PubMed Google Scholar * Wang Z, Sun R, Wang G, Chen Z, Li Y, Zhao Y, et al. SIRT3-mediated deacetylation of PRDX3 alleviates mitochondrial oxidative damage and apoptosis induced by
intestinal ischemia/reperfusion injury. Redox Biol. 2020;28:101343. Article CAS PubMed Google Scholar * Chua CC, Gao J, Ho YS, Xu X, Kuo IC, Chua KY, et al. Over-expression of a modified
bifunctional apoptosis regulator protects against cardiac injury and doxorubicin-induced cardiotoxicity in transgenic mice. Cardiovasc Res. 2009;81:20–7. Article CAS PubMed Google
Scholar * Wang Q, Zhou Y, Wang X, Evers BM. p27 Kip1 nuclear localization and cyclin-dependent kinase inhibitory activity are regulated by glycogen synthase kinase-3 in human colon cancer
cells. Cell Death Differ. 2008;15:908–19. Article CAS PubMed Google Scholar * Beach TE, Prag HA, Pala L, Logan A, Huang MM, Gruszczyk AV, et al. Targeting succinate dehydrogenase with
malonate ester prodrugs decreases renal ischemia reperfusion injury. Redox Biol. 2020;36:101640. Article CAS PubMed PubMed Central Google Scholar * Ding MJ, Fang HR, Zhang JK, Shi JH,
Yu X, Wen PH, et al. E3 ubiquitin ligase ring finger protein 5 protects against hepatic ischemia reperfusion injury by mediating phosphoglycerate mutase family member 5 ubiquitination.
Hepatology. 2022;76:94–111. Article CAS PubMed Google Scholar * Li Y, Xu J, Yu T, Zhu J, Xuan A, Liu X, et al. A labeling strategy for the three-dimensional recognition and analysis of
microvascular obstruction in ischemic stroke. Theranostics. 2023;13:403–16. Article CAS PubMed PubMed Central Google Scholar * Han JY, Li Q, Ma ZZ, Fan JY. Effects and mechanisms of
compound Chinese medicine and major ingredients on microcirculatory dysfunction and organ injury induced by ischemia/reperfusion. Pharm Ther. 2017;177:146–73. Article CAS Google Scholar *
Eltzschig HK, Eckle T. Ischemia and reperfusion-from mechanism to translation. Nat Med. 2011;17:1391–401. Article CAS PubMed Google Scholar * Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping
F, et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020;27:2635–50.
Article CAS PubMed PubMed Central Google Scholar * Mocanu MM, Baxter GF, Yellon DM. Caspase inhibition and limitation of myocardial infarct size: protection against lethal reperfusion
injury. Br J Pharm. 2000;130:197–200. Article CAS Google Scholar * Ikeda H, Suzuki Y, Suzuki M, Koike M, Tamura J, Tong J, et al. Apoptosis is a major mode of cell death caused by
ischaemia and ischaemia/reperfusion injury to the rat intestinal epithelium. Gut. 1998;42:530–7. Article CAS PubMed PubMed Central Google Scholar * Sies H. Oxidative stress: a concept
in redox biology and medicine. Redox Biol. 2015;4:180–3. Article CAS PubMed PubMed Central Google Scholar * Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and
disease: the therapeutic potential of Nrf2 activation. Mol Asp Med. 2011;32:234–46. Article CAS Google Scholar * Iqbal AJ, Fisher EA, Greaves DR. Inflammation—a critical appreciation of
the role of myeloid cells. Microbiol Spectr. 2016;4:1–17. Article CAS Google Scholar * Michopoulos V, Powers A, Gillespie CF, Ressler KJ, Jovanovic T. Inflammation in fear- and
anxiety-based disorders: PTSD, GAD, and beyond. Neuropsychopharmacology. 2017;42:254–70. Article CAS PubMed Google Scholar * Lu YY, Zhu CY, Ding YX, Wang B, Zhao SF, Lv J, et al.
Cepharanthine, a regulator of keap1-Nrf2, inhibits gastric cancer growth through oxidative stress and energy metabolism pathway. Cell Death Discov. 2023;9:450. Article CAS PubMed PubMed
Central Google Scholar * Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G, et al. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death
Discov. 2021;7:193. Article CAS PubMed PubMed Central Google Scholar * Wang J, Chen M, Wang S, Chu X, Ji H. Identification of phytogenic compounds with antioxidant action that protect
porcine intestinal epithelial cells from hydrogen peroxide induced oxidative damage. Antioxidants. 2022;11:2134. Article CAS PubMed PubMed Central Google Scholar * Bhattacharyya A,
Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev. 2014;94:329–54. Article CAS PubMed PubMed
Central Google Scholar * Perez-Asensio FJ, de la Rosa X, Jimenez-Altayo F, Gorina R, Martinez E, Messeguer A, et al. Antioxidant CR-6 protects against reperfusion injury after a transient
episode of focal brain ischemia in rats. J Cereb Blood Flow Metab. 2010;30:638–52. Article CAS PubMed Google Scholar * Han X, Yao W, Liu Z, Li H, Zhang ZJ, Hei Z, et al. Lipoxin A4
preconditioning attenuates intestinal ischemia reperfusion injury through Keap1/Nrf2 pathway in a lipoxin A4 receptor independent manner. Oxid Med Cell Longev. 2016;2016:9303606. Article
PubMed PubMed Central Google Scholar * Szabo C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharm Sci. 1998;19:287–98. Article CAS
PubMed Google Scholar * Cosyns SM, Shiva S, Lefebvre RA. Protective effect of exogenous nitrite in postoperative ileus. Br J Pharm. 2015;172:4864–74. Article CAS Google Scholar * Ku SK,
Han MS, Park EJ, Na DH, Bae JS. Exendin-4 inhibits endothelial protein C receptor shedding in vitro and in vivo. Pharm Res. 2014;84:18–25. Article CAS Google Scholar * Collange O,
Charles AL, Lavaux T, Noll E, Bouitbir J, Zoll J, et al. Compartmentalization of inflammatory response following gut ischemia reperfusion. Eur J Vasc Endovasc Surg. 2015;49:60–65. Article
CAS PubMed Google Scholar * Bertoni S, Arcaro V, Vivo V, Rapalli A, Tognolini M, Cantoni AM, et al. Suppression of inflammatory events associated to intestinal ischemia-reperfusion by
5-HT1A blockade in mice. Pharm Res. 2014;81:17–25. Article CAS Google Scholar * Kim JH, Kim J, Chun J, Lee C, Im JP, Kim JS. Role of iRhom2 in intestinal ischemia-reperfusion-mediated
acute lung injury. Sci Rep. 2018;8:3797. Article ADS PubMed PubMed Central Google Scholar * Huang T, Cao Y, Wang H, Wang Q, Ji J, Sun X, et al. Circular RNA YAP1 acts as the sponge of
microRNA-21-5p to secure HK-2 cells from ischaemia/reperfusion-induced injury. J Cell Mol Med. 2020;24:4707–15. Article CAS PubMed PubMed Central Google Scholar * Dong J, Liang W, Wang
T, Sui J, Wang J, Deng Z, et al. Saponins regulate intestinal inflammation in colon cancer and IBD. Pharm Res. 2019;144:66–72. Article CAS Google Scholar * Arifa RDN, de Paula TP, Lima
RL, Brito CB, Andrade MER, Cardoso VN, et al. Anti-inflammatory and antioxidant effects of the nanocomposite Fullerol decrease the severity of intestinal inflammation induced by gut ischemia
and reperfusion. Eur J Pharm. 2021;898:173984. Article CAS Google Scholar * Feng D, Yao J, Wang G, Li Z, Zu G, Li Y, et al. Inhibition of p66Shc-mediated mitochondrial apoptosis via
targeting prolyl-isomerase Pin1 attenuates intestinal ischemia/reperfusion injury in rats. Clin Sci. 2017;131:759–73. Article CAS Google Scholar * Boengler K, Bornbaum J, Schluter KD,
Schulz R. P66shc and its role in ischemic cardiovascular diseases. Basic Res Cardiol. 2019;114:29. Article PubMed Google Scholar * Feng D, Wang Z, Zhao Y, Li Y, Liu D, Chen Z, et al.
circ-PRKCB acts as a ceRNA to regulate p66Shc-mediated oxidative stress in intestinal ischemia/reperfusion. Theranostics. 2020;10:10680–96. Article CAS PubMed PubMed Central Google
Scholar * Turan I, Ozacmak HS, Ozacmak VH, Barut F, Arasli M. Agmatine attenuates intestinal ischemia and reperfusion injury by reducing oxidative stress and inflammatory reaction in rats.
Life Sci. 2017;189:23–8. Article CAS PubMed Google Scholar * Hu Y, Mao Z, Xu L, Yin L, Tao X, Tang Z, et al. Protective effect of dioscin against intestinal ischemia/reperfusion injury
via adjusting miR-351-5p-mediated oxidative stress. Pharm Res. 2018;137:56–63. Article CAS Google Scholar * Li LX, Yin LH, Gao M, Xu LN, Qi Y, Peng JY. MiR-23a-5p exacerbates intestinal
ischemia-reperfusion injury by promoting oxidative stress via targeting PPAR alpha. Biochem Pharm. 2020;180:114194. Article CAS PubMed Google Scholar * Wang Y, Wen J, Almoiliqy M, Wang
Y, Liu Z, Yang X, et al. Sesamin protects against and ameliorates rat intestinal ischemia/reperfusion injury with involvement of activating Nrf2/HO-1/NQO1 signaling pathway. Oxid Med Cell
Longev. 2021;2021:5147069. Article PubMed PubMed Central Google Scholar * Dou X, Chen L, Lei M, Zellmer L, Jia Q, Ling P, et al. Evaluating the remote control of programmed cell death,
with or without a compensatory cell proliferation. Int J Biol Sci. 2018;14:1800–12. Article PubMed PubMed Central Google Scholar * Yu J, Zhong B, Xiao Q, Du L, Hou Y, Sun HS, et al.
Induction of programmed necrosis: a novel anti-cancer strategy for natural compounds. Pharm Ther. 2020;214:107593. Article CAS Google Scholar * Mo K, Chu Y, Liu Y, Zheng G, Song K, Song
Q, et al. Targeting hnRNPC suppresses thyroid follicular epithelial cell apoptosis and necroptosis through m(6)A-modified ATF4 in autoimmune thyroid disease. Pharm Res. 2023;196:106933.
Article CAS Google Scholar * Guneli E, Cavdar Z, Islekel H, Sarioglu S, Erbayraktar S, Kiray M, et al. Erythropoietin protects the intestine against ischemia/ reperfusion injury in rats.
Mol Med. 2007;13:509–17. Article CAS PubMed PubMed Central Google Scholar * Du L, Yu Y, Ma H, Lu X, Ma L, Jin Y, et al. Hypoxia enhances protective effect of placental-derived
mesenchymal stem cells on damaged intestinal epithelial cells by promoting secretion of insulin-like growth factor-1. Int J Mol Sci. 2014;15:1983–2002. Article PubMed PubMed Central
Google Scholar * Ho CJ, Ko HJ, Liao TS, Zheng XR, Chou PH, Wang LT, et al. Severe cellular stress activates apoptosis independently of p53 in osteosarcoma. Cell Death Discov. 2021;7:275.
Article CAS PubMed PubMed Central Google Scholar * Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell
Biol. 2007;8:741–52. Article CAS PubMed Google Scholar * Kaminskyy VO, Zhivotovsky B. Free radicals in cross talk between autophagy and apoptosis. Antioxid Redox Signal. 2014;21:86–102.
Article CAS PubMed Google Scholar * Fleisher TA. Apoptosis. Ann Allergy Asthma Immunol. 1997;78:245–9. Article CAS PubMed Google Scholar * Hotchkiss RS, Strasser A, McDunn JE,
Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83. Article CAS PubMed PubMed Central Google Scholar * Sauler M, Bazan IS, Lee PJ. Cell death in the lung: the apoptosis-necroptosis
axis. Annu Rev Physiol. 2019;81:375–402. Article CAS PubMed Google Scholar * Liu DQ, Chen SP, Sun J, Wang XM, Chen N, Zhou YQ, et al. Berberine protects against ischemia-reperfusion
injury: a review of evidence from animal models and clinical studies. Pharm Res. 2019;148:104385. Article CAS Google Scholar * Zhang X, Wu J, Liu Q, Li X, Li S, Chen J, et al. mtDNA-STING
pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis. 2020;11:1050. Article PubMed PubMed Central Google Scholar * Cao Y, Huang W,
Wu F, Shang J, Ping F, Wang W, et al. ZFP36 protects lungs from intestinal I/R-induced injury and fibrosis through the CREBBP/p53/p21/Bax pathway. Cell Death Dis. 2021;12:685. Article CAS
PubMed PubMed Central Google Scholar * Zhang Q, Liu XM, Hu Q, Liu ZR, Liu ZY, Zhang HG, et al. Dexmedetomidine inhibits mitochondria damage and apoptosis of enteric glial cells in
experimental intestinal ischemia/reperfusion injury via SIRT3-dependent PINK1/HDAC3/p53 pathway. J Transl Med. 2021;19:463. Article CAS PubMed PubMed Central Google Scholar * Zheng L,
Han X, Hu Y, Zhao X, Yin L, Xu L, et al. Dioscin ameliorates intestinal ischemia/reperfusion injury via adjusting miR-351-5p/MAPK13-mediated inflammation and apoptosis. Pharm Res.
2019;139:431–9. Article CAS Google Scholar * Amaral FA, Fagundes CT, Guabiraba R, Vieira AT, Souza AL, Russo RC, et al. The role of macrophage migration inhibitory factor in the cascade
of events leading to reperfusion-induced inflammatory injury and lethality. Am J Pathol. 2007;171:1887–93. Article CAS PubMed PubMed Central Google Scholar * Yu P, Zhang X, Liu N, Tang
L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6:128. Article PubMed PubMed Central Google Scholar * Bertheloot D, Latz E, Franklin BS.
Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18:1106–21. Article CAS PubMed PubMed Central Google Scholar * Tang R, Xu J, Zhang B, Liu
J, Liang C, Hua J, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. 2020;13:110. Article PubMed PubMed Central Google Scholar * Jia Y, Cui R, Wang
C, Feng Y, Li Z, Tong Y, et al. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020;32:101534. Article CAS
PubMed PubMed Central Google Scholar * Wang F, Gu L, Wang Y, Sun D, Zhao Y, Meng Q, et al. MicroRNA-122a aggravates intestinal ischemia/reperfusion injury by promoting pyroptosis via
targeting EGFR-NLRP3 signaling pathway. Life Sci. 2022;307:120863. Article CAS PubMed Google Scholar * Guo S, Fu Y, Xiong S, Lv J. Corilagin protects the acute lung injury by
ameliorating the apoptosis pathway. Biomed Pharmacother. 2017;95:1743–8. Article CAS PubMed Google Scholar * Li W, Yang K, Li B, Wang Y, Liu J, Chen D, et al. Corilagin alleviates
intestinal ischemia/reperfusion-induced intestinal and lung injury in mice via inhibiting NLRP3 inflammasome activation and pyroptosis. Front Pharm. 2022;13:1060104. Article CAS Google
Scholar * Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:588–606. Article CAS PubMed
Google Scholar * Wang L, Hauenstein AV. The NLRP3 inflammasome: mechanism of action, role in disease and therapies. Mol Asp Med. 2020;76:100889. Article CAS Google Scholar * Cui Y,
Zhang Z, Zhou X, Zhao Z, Zhao R, Xu X, et al. Microglia and macrophage exhibit attenuated inflammatory response and ferroptosis resistance after RSL3 stimulation via increasing Nrf2
expression. J Neuroinflammation. 2021;18:249. Article CAS PubMed PubMed Central Google Scholar * Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ
oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92. Article ADS CAS PubMed PubMed Central Google Scholar * Jiang X, Stockwell BR, Conrad M.
Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82. Article PubMed PubMed Central Google Scholar * Liang C, Zhang X, Yang M, Dong X. Recent
progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31:e1904197. Article PubMed Google Scholar * Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to
iron out cancer. Cancer Cell. 2019;35:830–49. Article CAS PubMed Google Scholar * Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, et al. Ischemia-induced ACSL4 activation contributes to
ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019;26:2284–99. Article CAS PubMed PubMed Central Google Scholar * Xu S, He Y, Lin L, Chen P,
Chen M, Zhang S. The emerging role of ferroptosis in intestinal disease. Cell Death Dis. 2021;12:289. Article PubMed PubMed Central Google Scholar * Dong H, Qiang Z, Chai D, Peng J, Xia
Y, Hu R, et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging. 2020;12:12943–59. Article CAS
PubMed PubMed Central Google Scholar * Dong H, Xia Y, Jin S, Xue C, Wang Y, Hu R, et al. Nrf2 attenuates ferroptosis-mediated IIR-ALI by modulating TERT and SLC7A11. Cell Death Dis.
2021;12:1027. Article CAS PubMed PubMed Central Google Scholar * Li M, He Z, Zhong H, Sun W, Ye M, Zhou Y. A novel multi-components hierarchical porous composite prepared from solid
wastes for benzohydroxamic acid degradation. J Colloid Interface Sci. 2023;630:714–26. Article CAS PubMed Google Scholar * Wan J, Ren H, Wang J. Iron toxicity, lipid peroxidation and
ferroptosis after intracerebral haemorrhage. Stroke Vasc Neurol. 2019;4:93–5. Article PubMed PubMed Central Google Scholar * Wang YH, Yan ZZ, Luo SD, Hu JJ, Wu M, Zhao J, et al. Gut
microbiota-derived succinate aggravates acute lung injury after intestinal ischaemia/reperfusion in mice. Eur Respir J. 2023;61:2200840. Article CAS PubMed Google Scholar * Huang W, Yan
Y, Wu M, Hu J, Zhao J, Chen X, et al. Preoperative fasting confers protection against intestinal ischaemia/reperfusion injury by modulating gut microbiota and their metabolites in a mouse
model. Br J Anaesth. 2022;128:501–12. Article PubMed Google Scholar * Deng F, Zhao BC, Yang X, Lin ZB, Sun QS, Wang YF, et al. The gut microbiota metabolite capsiate promotes Gpx4
expression by activating TRPV1 to inhibit intestinal ischemia reperfusion-induced ferroptosis. Gut Microbes. 2021;13:1–21. Article PubMed Google Scholar * Cai M, Ma Y, Zhang W, Wang S,
Wang Y, Tian L, et al. Apigenin-7-O-beta-D-(-6”-p-coumaroyl)-glucopyranoside treatment elicits neuroprotective effect against experimental ischemic stroke. Int J Biol Sci. 2016;12:42–52.
Article CAS PubMed PubMed Central Google Scholar * Feng YD, Ye W, Tian W, Meng JR, Zhang M, Sun Y, et al. Old targets, new strategy:
apigenin-7-O-beta-d-(-6”-p-coumaroyl)-glucopyranoside prevents endothelial ferroptosis and alleviates intestinal ischemia-reperfusion injury through HO-1 and MAO-B inhibition. Free Radic
Biol Med. 2022;184:74–88. Article CAS PubMed Google Scholar * Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell.
2022;82:2215–27. Article CAS PubMed PubMed Central Google Scholar * Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis.
2020;11:88. Article PubMed PubMed Central Google Scholar * Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25. Article
CAS PubMed Google Scholar * Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. Article CAS PubMed Google Scholar * Glick D, Barth S, Macleod
KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. Article CAS PubMed PubMed Central Google Scholar * Levine B, Kroemer G. Biological functions of autophagy
genes: a disease perspective. Cell. 2019;176:11–42. Article CAS PubMed PubMed Central Google Scholar * Kim KH, Lee MS. Autophagy—a key player in cellular and body metabolism. Nat Rev
Endocrinol. 2014;10:322–37. Article CAS PubMed Google Scholar * Zhenzhen L, Wenting L, Jianmin Z, Guangru Z, Disheng L, Zhiyu Z, et al. miR-146a-5p/TXNIP axis attenuates intestinal
ischemia-reperfusion injury by inhibiting autophagy via the PRKAA/mTOR signaling pathway. Biochem Pharm. 2022;197:114839. Article PubMed Google Scholar * Wen J, Xu B, Sun Y, Lian M, Li Y,
Lin Y, et al. Paeoniflorin protects against intestinal ischemia/reperfusion by activating LKB1/AMPK and promoting autophagy. Pharm Res. 2019;146:104308. Article CAS Google Scholar * Li
Y, Zhang P, Zhang J, Bao W, Li J, Wei Y, et al. Role of autophagy inducers and inhibitors in intestinal barrier injury induced by intestinal ischemia-reperfusion (I/R). J Immunol Res.
2022;2022:9822157. Article PubMed PubMed Central Google Scholar * Li Z, Wang G, Feng D, Zu G, Li Y, Shi X, et al. Targeting the miR-665-3p-ATG4B-autophagy axis relieves inflammation and
apoptosis in intestinal ischemia/reperfusion. Cell Death Dis. 2018;9:483. Article PubMed PubMed Central Google Scholar * Li S, Zhou Y, Gu X, Zhang X, Jia Z. NLRX1/FUNDC1/NIPSNAP1-2 axis
regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 2021;54:e12986. Article CAS PubMed PubMed Central Google Scholar * Dusabimana T, Kim SR, Kim HJ,
Park SW, Kim H. Nobiletin ameliorates hepatic ischemia and reperfusion injury through the activation of SIRT-1/FOXO3a-mediated autophagy and mitochondrial biogenesis. Exp Mol Med.
2019;51:1–16. Article CAS PubMed Google Scholar * Shi D, Li X, Li D, Zhao Q, Shen Y, Yan H, et al. Oral administration of paeoniflorin attenuates allergic contact dermatitis by
inhibiting dendritic cell migration and Th1 and Th17 differentiation in a mouse model. Int Immunopharmacol. 2015;25:432–9. Article CAS PubMed Google Scholar * Ma Z, Chu L, Liu H, Wang W,
Li J, Yao W, et al. Beneficial effects of paeoniflorin on non-alcoholic fatty liver disease induced by high-fat diet in rats. Sci Rep. 2017;7:44819. Article ADS PubMed PubMed Central
Google Scholar * Frankel LB, Wen J, Lees M, Hoyer-Hansen M, Farkas T, Krogh A, et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011;30:4628–41. Article CAS PubMed PubMed
Central Google Scholar * Snaka T, Bekkar A, Desponds C, Prevel F, Claudinot S, Isorce N, et al. Sex-biased control of inflammation and metabolism by a mitochondrial nod-like receptor.
Front Immunol. 2022;13:882867. Article CAS PubMed PubMed Central Google Scholar * Hu B, Ding GY, Fu PY, Zhu XD, Ji Y, Shi GM, et al. NOD-like receptor X1 functions as a tumor suppressor
by inhibiting epithelial-mesenchymal transition and inducing aging in hepatocellular carcinoma cells. J Hematol Oncol. 2018;11:28. Article PubMed PubMed Central Google Scholar * Liu K,
Cai Z, Zhang Q, He J, Cheng Y, Wei S, et al. Determination of significant parameters in remote ischemic postconditioning for ischemic stroke in experimental models: a systematic review and
meta-analysis study. CNS Neurosci Ther. 2022;28:1492–508. Article PubMed PubMed Central Google Scholar * Li Y, Luo Y, Li B, Niu L, Liu J, Duan X. miRNA-182/Deptor/mTOR axis regulates
autophagy to reduce intestinal ischaemia/reperfusion injury. J Cell Mol Med. 2020;24:7873–83. Article CAS PubMed PubMed Central Google Scholar * Zhu Q, He G, Wang J, Wang Y, Chen W.
Pretreatment with the ALDH2 agonist Alda-1 reduces intestinal injury induced by ischaemia and reperfusion in mice. Clin Sci. 2017;131:1123–36. Article CAS Google Scholar * Zhao XH, Yang
T, Zheng MY, Zhao P, An LY, Qi YX, et al. Cystathionine gamma-lyase (Cth) induces efferocytosis in macrophages via ERK1/2 to modulate intestinal barrier repair. Cell Commun Signal.
2023;21:17. Article CAS PubMed PubMed Central Google Scholar * Zhang X, Wu J, Liu Q, Li X, Li S, Chen J, et al. mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in
intestinal ischemia reperfusion. Cell Death Dis. 2020;11:1050. Article CAS PubMed PubMed Central Google Scholar * Hu Q, Ren J, Li G, Wu J, Wu X, Wang G, et al. The mitochondrially
targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018;9:403. Article PubMed PubMed
Central Google Scholar * Fusco R, Cordaro M, Siracusa R, Peritore AF, Gugliandolo E, Genovese T, et al. Consumption of Anacardium Occidentale L. (cashew nuts) inhibits oxidative stress
through modulation of the Nrf2/HO-1 and NF-kB pathways. Molecules. 2020;25:4426. Article CAS PubMed PubMed Central Google Scholar * Fan X, Du J, Wang MH, Li JM, Yang B, Chen Y, et al.
Irisin contributes to the hepatoprotection of dexmedetomidine during intestinal ischemia/reperfusion. Oxid Med Cell Longev. 2019;2019:7857082. Article PubMed PubMed Central Google Scholar
* Li L, Tong A, Zhang Q, Wei Y, Wei X. The molecular mechanisms of MLKL-dependent and MLKL-independent necrosis. J Mol Cell Biol. 2021;13:3–14. Article CAS PubMed Google Scholar * Zhou
P, Zhang S, Wang M, Zhou J. The induction mechanism of ferroptosis, necroptosis, and pyroptosis in inflammatory bowel disease, colorectal cancer, and intestinal injury. Biomolecules.
2023;13:820. Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We would like to acknowledge the reviewers for their helpful comments on this paper.
FUNDING This study was financially supported by the Fundamental Research Funds for the Central Universities (2023-JYB-JBQN-051), and the Talent Cultivation Project of Beijing University of
Chinese Medicine (JZPY202206). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * School of Chinese Materia Medica, Beijing University of Chinese Medicine, 100029, Beijing, China Fei Wang,
Huiming Huang, Xuejiao Wei, Peng Tan & Zhuguo Wang * Modern Research Center for Traditional Chinese Medicine, Beijing Research Institute of Chinese Medicine, Beijing University of
Chinese Medicine, 100029, Beijing, China Fei Wang, Huiming Huang, Xuejiao Wei, Peng Tan, Zhuguo Wang & Zhongdong Hu Authors * Fei Wang View author publications You can also search for
this author inPubMed Google Scholar * Huiming Huang View author publications You can also search for this author inPubMed Google Scholar * Xuejiao Wei View author publications You can also
search for this author inPubMed Google Scholar * Peng Tan View author publications You can also search for this author inPubMed Google Scholar * Zhuguo Wang View author publications You can
also search for this author inPubMed Google Scholar * Zhongdong Hu View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS FW: conceptualization,
literature search, writing—original draft. HH, XW, PT, and ZW: writing and revision of the paper. ZH: conceptualization, supervision, funding acquisition, writing—review and editing. All the
authors have read and approved the final version of the manuscript. CORRESPONDING AUTHOR Correspondence to Zhongdong Hu. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no
competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Wang, F., Huang,
H., Wei, X. _et al._ Targeting cell death pathways in intestinal ischemia-reperfusion injury: a comprehensive review. _Cell Death Discov._ 10, 112 (2024).
https://doi.org/10.1038/s41420-024-01891-x Download citation * Received: 27 November 2023 * Revised: 21 February 2024 * Accepted: 26 February 2024 * Published: 04 March 2024 * DOI:
https://doi.org/10.1038/s41420-024-01891-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative