Sprr1a is a key downstream effector of mir-150 during both maladaptive cardiac remodeling in mice and human cardiac fibroblast activation
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT MicroRNA-150 (miR-150) is conserved between rodents and humans, is significantly downregulated during heart failure (HF), and correlates with patient outcomes. We previously
reported that miR-150 is protective during myocardial infarction (MI) in part by decreasing cardiomyocyte (CM) apoptosis and that proapoptotic small proline-rich protein 1a (_Sprr1a_) is a
direct CM target of miR-150. We also showed that _Sprr1a_ knockdown in mice improves cardiac dysfunction and fibrosis post-MI and that _Sprr1a_ is upregulated in pathological mouse cardiac
fibroblasts (CFs) from ischemic myocardium. However, the direct functional relationship between miR-150 and SPRR1A during both post-MI remodeling in mice and human CF (HCF) activation was
not established. Here, using a novel miR-150 knockout;_Sprr1a_-hypomorphic (_Sprr1a__hypo/hypo_) mouse model, we demonstrate that _Sprr1a_ knockdown blunts adverse post-MI effects caused by
miR-150 loss. Moreover, HCF studies reveal that _SPRR1A_ is upregulated in hypoxia/reoxygenation-treated HCFs and is downregulated in HCFs exposed to the cardioprotective β-blocker
carvedilol, which is inversely associated with miR-150 expression. Significantly, we show that the protective roles of miR-150 in HCFs are directly mediated by functional repression of
profibrotic _SPRR1A_. These findings delineate a pivotal functional interaction between miR-150 and SPRR1A as a novel regulatory mechanism pertinent to CF activation and ischemic HF. SIMILAR
CONTENT BEING VIEWED BY OTHERS CIRCNSD1 PROMOTES CARDIAC FIBROSIS THROUGH TARGETING THE MIR-429-3P/SULF1/WNT/Β-CATENIN SIGNALING PATHWAY Article 17 May 2024 CIRCULAR _PVT1_ PROMOTES CARDIAC
FIBROBLAST ACTIVATION INTERACTING WITH MIR-30A-5P AND MIR-125B-5P Article Open access 21 April 2025 THE DOUBLE FACE OF MIR-320: CARDIOMYOCYTES-DERIVED MIR-320 DETERIORATED WHILE
FIBROBLASTS-DERIVED MIR-320 PROTECTED AGAINST HEART FAILURE INDUCED BY TRANSVERSE AORTIC CONSTRICTION Article Open access 18 February 2021 INTRODUCTION Controlling microRNA (miRNA or miR)
biogenesis in the heart is an important underlying mechanism of heart failure (HF) [1,2,3,4,5]. Intriguingly, novel miR therapies are being investigated in clinical trials for other diseases
[6,7,8,9] and more recently for HF [10]. We reported that miR-150 is upregulated by the β-blocker carvedilol (Carv), which acts through β-arrestin1-mediated β1-adrenergic receptor (β1AR)
protective signaling [11]. Significantly, using a systemic miR-150 knockout (KO) mouse model, we also showed that miR-150 plays a vital Carv/β1AR/β-arrestin1-mediated protective role in
myocardial infarction (MI) in part by decreasing cardiomyocyte (CM) apoptosis [12]. More recently, we demonstrated that cardiac-specific miR-150 conditional KO (cKO) mice exhibit enhanced
apoptosis and maladaptive post-MI remodeling [13]. Notably, cardiac-specific overexpression of miR-150 attenuated transverse aortic constriction (TAC)-induced cardiac dysfunction [14]
whereas miR-150 loss caused a higher degree of cardiac fibrosis after TAC. MiR-150 was also downregulated in cardiac fibroblasts (CFs), not CMs isolated from TAC mice [15]. Moreover, miR-150
inhibited mouse CF activation in vitro [15]. Interestingly, circulating or cardiac miR-150 is downregulated in patients with multiple cardiovascular diseases (CVDs) [16,17,18,19] and mouse
models of HF [12, 14, 20]. MiR-150 is conserved between rodents and humans and is significantly associated with HF severity and outcome in humans [21]. Collectively, previous studies support
the clinical relevance and potential therapeutic application of miR-150 in HF; however, the detailed mechanisms by which miR-150 modulates HF remain elusive. Small proline-rich protein 1a
(SPRR1A) is induced by stress and is highly conserved. SPRR1A is a substrate of transglutaminase (TGase) I/II-catalyzed crosslinking reactions in forming the keratinocyte envelope [22]. We
showed that CMs with _Sprr1a_ knockdown are protected against apoptosis in the simulated ischemia/reperfusion (sI/R: hypoxia/reoxygenation [H/R]) condition [13]. Adenovirus-mediated ectopic
overexpression of _Sprr1a_ promoted cardiac fibrosis in vivo after TAC whereas protecting CMs and isolated hearts against 2-deoxyglucose and ex vivo I/R [23]. Moreover, _Sprr1a_
overexpression did not affect CM survival after reactive oxygen species treatment or serum deprivation [23]. This prior study indicated both the potential stress-dependent effects of
_Sprr1a_ and the requirement of in vivo genetic loss-of-function approaches to define the role of _Sprr1a_ in the heart. Interestingly, we recently demonstrated that _Sprr1a_-hypomorphic
(_Sprr1a__hypo/hypo_) mice are protected against MI [13]. We also reported that _Sprr1a_ is upregulated in CMs isolated from mouse hearts post-MI [13], which is consistent with a report on
mouse hearts after TAC [23]. Moreover, we showed that left ventricular (LV) _SPRR1A_ is upregulated in patients with HF [13] in agreement with mouse studies showing _Sprr1a_ upregulation in
myocardial injury [24] and renal I/R injury [25]. Notably, we identified proapoptotic _Sprr1a_ as a novel direct and functional target of miR-150 in CMs [13]. We also showed that _Sprr1a_ is
downregulated by Carv in hearts and CMs [13] concurrent with miR-150 upregulation [11]. Given that rodent and human genes of SPRR1A have almost identical genomic organization and
exon/intron sizes [26] and have at least one miR-150 binding site, the regulation of SPRR1A by miR-150 and their roles might be conserved. Importantly, we also showed that _Sprr1a_ knockdown
in mice improves fibrosis post-MI and that _Sprr1a_ is upregulated in mouse CFs isolated from ischemic myocardium [13]. A recent proteomic study in CFs also noted that SPRR1A levels are
significantly higher in infarct CFs than remote CFs during MI [27]. These previous findings indicate a possible role of _Sprr1a_ in CFs; however, whether _Sprr1a_ is functionally regulated
by miR-150 in HF and human CF (HCF) activation remains unknown. Using a novel miR-150 KO;_Sprr1a_-hypomorphic (_Sprr1a__hypo/hypo_) mouse model and primary HCFs, we demonstrate here that (i)
_Sprr1a_ knockdown alleviates cardiac dysfunction, damage, apoptosis, and fibrosis after MI mediated by miR-150 deletion; (ii) _Sprr1a_ is upregulated in HCFs subjected to H/R whereas its
expression is downregulated in HCFs by Carv, which is inversely associated with the expression of miR-150; and (iii) the protective actions of miR-150 in HCFs are mediated by the functional
repression of profibrotic _SPRR1A_. These data directly establish the functional relationship between miR-150 and _Sprr1a_ during both post-MI fibrotic remodeling in mice and HCF activation.
Our novel findings suggest that profibrotic SPRR1A is a crucial downstream effector of miR-150 in repressing CF activation and maladaptive cardiac remodeling. The miR-150/SPRR1A axis,
therefore, may be considered a novel therapeutic target for ameliorating ischemic heart disease and pathological fibrosis. MATERIALS AND METHODS MIR-150 KNOCKOUT AND HYPOMORPHIC _SPRR1A_
MUTANT MICE (_SPRR1A_ _HYPO/HYPO_) AS WELL AS THE GENERATION OF MIR-150 KO;_SPRR1A_ _HYPO/HYPO_ MICE Systemic miR-150 KO mice were purchased from the Jackson Laboratory (007750), and their
cardiac phenotypes post-MI were reported in our previous study [12]. _Sprr1a__hypo/+_ were obtained from the Mutant Mouse Resource & Research Centers (RRID: MMRRC_049856-UCD). We
previously described the detailed methods regarding this mouse line, including the targeting strategy, generation of _Sprr1a__hypo/hypo_ mice, and genotyping strategy [13]. We also reported
the cardiac phenotypes of _Sprr1a__hypo/hypo_ mice after MI in a previous study [13]. In the current study, _Sprr1a__hypo/hypo_ mice were bred with miR-150 KO mice to generate the novel
miR-150 KO;_Sprr1a__hypo/hypo_ mouse line. All mice were maintained on a C57BL/6J background, and wild-type (WT) littermates were used as controls. ETHICS COMMITTEE APPROVAL The animal
experiments conducted as a part of this study complied with the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Mice were euthanized
by thoracotomy under 1–4% inhaled isoflurane. All experiments with mice were performed according to the protocols approved by the Institutional Animal Care and Use Committee at the Indiana
University School of Medicine (approval reference #21189). 8–16 week-old C57BL/6J mice of both sexes were used for this study. Genotype- and sex-matched mice were randomly assigned to
experimental groups to mitigate the cage effect. The genotypes of the animals were masked for researchers until the end of the analysis. STATISTICS Data are presented as the mean ± SEM
(except Fig. 1 using SD because no clear variation bars are shown otherwise) from independent experiments with different biological samples per group. Triplicate experiments were performed
for all biochemical and cell biology studies. The number of in vitro biological samples per group was 3–6. The number of mouse samples per group was 3–18. The exact sample size for each
experimental group/condition is given as a number in the figure/table legend. To ensure the robustness of the data and to allow the direct evaluation of the distribution of the data, we
present graphical data as scatter/dot plots. Normality was assessed with the Kolmogorov-Smirnov test. The following statistical tests were used: unpaired two-tailed t-test for comparisons
between 2 groups, one-way ANOVA with Tukey’s multiple comparison test for multiple groups, two-way ANOVA with Tukey’s multiple comparison test for comparisons between 2 groups with different
treatments, and two-way repeated-measures ANOVA with Bonferroni’s post hoc test for 2 groups over time. The unpaired 2-tailed t-test was based on assumed normal distributions. A _P_ value
< 0.05 was considered statistically significant. _P_ values are indicated as follows: *,#,or§_P_ < 0.05, **,##,or§§_P_ < 0.01, and ***,###,or§§§_P_ < 0.001. RESULTS SPRR1A
KNOCKDOWN IN MIR-150 KO MICE LARGELY CORRECTS CARDIAC DYSFUNCTION MEDIATED BY MIR-150 DELETION _Sprr1a_ is a direct target of miR-150 in vitro, miR-150 acts as a gatekeeper of CM survival in
part by inhibiting proapoptotic _Sprr1a_ [13], and their correlative cardiac actions are shown [12, 13]; but an in vivo functional relationship between miR-150 and _Sprr1a_ in the heart has
not been established. To directly investigate their in vivo functional interaction in the heart, we generated a novel miR-150 KO;_Sprr1a__hypo/hypo_ mouse line by breeding miR-150 KO mice
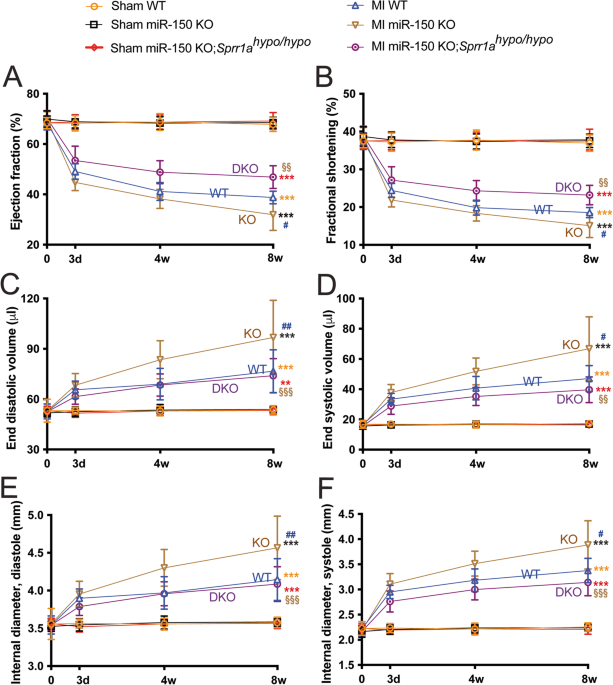
with _Sprr1a__hypo/hypo_ mice. We first conducted permanent ligation of the left anterior descending (LAD) artery in mice to induce MI. Consistent with a previous report [12], we observe
that miR-150 KO mice exhibit normal cardiac function at baseline (Supplementary Table 1 and Fig. 1) but respond differently to MI. Cardiac function is significantly compromised in
miR-150-null mice following MI. First, MI significantly worsens the cardiac function of miR-150 KO mice at 3 days as indicated by a decreased ejection fraction (EF), fractional shortening
(FS), diastolic left ventricular anterior wall thickness (LVAW), and systolic left ventricular posterior wall thickness (LVPW) as well as an increase in end-systolic volume (ESV) and
systolic left ventricular internal diameter (LVID) compared to those of WT controls (Supplementary Table 2 and Fig. 1). MiR-150 KO mice also display impaired cardiac function at 4 weeks
post-MI, shown by a significant decrease in EF, FS, diastolic LVPW, and systolic LVPW as well as a significant increase in end-diastolic volume (EDV), ESV, diastolic LVID, and systolic LVID
(Supplementary Table 3 and Fig. 1). MI also causes augmented cardiac dysfunction in miR-150 KO mice at 8 weeks as evidenced by a significant decrease in EF, FS, diastolic LVAW, diastolic
LVPW, and systolic LVPW as well as a significant increase in EDV, ESV, diastolic LVID, and systolic LVID (Supplementary Table 4 and Fig. 1). In contrast, WT controls show less functional
impairment at 4 weeks (Supplementary Table 3 and Fig. 1) and 8 weeks following MI (Supplementary Table 4 and Fig. 1). We next show that miR-150 KO;_Sprr1a__hypo/hypo_ mouse hearts are
functionally normal at baseline (Supplementary Table 1 and Fig. 1). However, a significant improvement in cardiac function at 3 days after MI is observed in miR-150 KO;_Sprr1a__hypo/hypo_
mice compared to miR-150 KO mice, indicated by an increase in cardiac output (CO), EF, FS, and diastolic LVAW as well as a decrease in EDV, ESV, diastolic LVID, and systolic LVID
(Supplementary Table 2 and Fig. 1). MiR-150 KO;_Sprr1a__hypo/hypo_ mice also display enhanced cardiac function at 4 weeks post-MI as evidenced by a significant increase in EF, FS, diastolic
LVAW, systolic LVAW, diastolic LVPW, and systolic LVPW as well as a significant decrease in EDV, ESV, diastolic LVID, and systolic LVID (Supplementary Table 3 and Fig. 1) compared to those
of miR-150 KO mice. Last, we show improved cardiac function in miR-150 KO;_Sprr1a__hypo/hypo_ mice at 8 weeks post-MI compared to miR-150 KO mice as shown by a significant increase in CO,
EF, FS, heart rate (HR), diastolic LVAW, systolic LVAW, and systolic LVPW as well as a significant decrease in EDV, ESV, diastolic LVID, and systolic LVID (Supplementary Table 4 and Fig. 1).
Our morphometric data also show that miR-150 KO;_Sprr1a__hypo/hypo_ mice have a significant decrease in the ratio of heart weight/body weight (HW/BW) and the ratio of left ventricle
weight/body weight (LVW/BW) at 8 weeks after MI compared to miR-150 KO controls (Supplementary Table 4). Notably, we do not observe any difference in post-MI mortality between groups
(Supplementary Tables 1, 3, and 4: see n for animal numbers per each group at week 0, week 4, and week 8 after MI). SUSTAINED SPRR1A KNOCKDOWN ALLEVIATES CARDIAC DAMAGE, INFLAMMATION, AND
APOPTOSIS POST-MI MEDIATED BY MIR-150 LOSS We previously reported that miR-150 KO mice display excessive maladaptive post-MI remodeling, such as cardiac damage, inflammation, and apoptosis
[12]. To determine whether repression of _Sprr1a_ mediates the major functions of miR-150 in vivo, we employed miR-150 KO;_Sprr1a__hypo/hypo_ mice and assessed post-MI remodeling compared to
that of miR-150 KO controls. We find that miR-150 KO;_Sprr1a__hypo/hypo_ hearts exhibit a decrease in the loss of normal architecture and cellular integrity (Fig. 2A) as well as decreased
mRNA levels of fetal _Nppa_ (Fig. 2B) after 8 weeks of MI compared to miR-150 KO hearts. We next examined whether an improved cardiac inflammatory cell (CI) response contributes to the
decreased disorganized structure in miR-150 KO;_Sprr1a__hypo/hypo_ hearts post-MI. Notably, inflammatory _Il-6_, _Tnf-α_, and _Ptprc_ are also downregulated in miR-150 KO;_Sprr1a__hypo/hypo_
hearts (Fig. 2C, D and Supplementary Fig. 1) compared to miR-150 KO hearts post-MI. Finally, we find that miR-150 KO;_Sprr1a__hypo/hypo_ hearts contain significantly lower numbers of
cleaved caspase-3-positive cells (Fig. 3A, B), indicating decreased apoptosis in miR-150 KO;_Sprr1a__hypo/hypo_ hearts. Our data further show that miR-150 KO;_Sprr1a__hypo/hypo_ hearts have
decreased mRNA levels of apoptotic _P53_, _Bak1_, and _Bax_ (Fig. 3C–E) compared to levels in miR-150 KO hearts. Altogether, our data suggest that sustained _Sprr1a_ downregulation
ameliorates adverse post-MI remodeling caused by miR-150 deletion and that miR-150 is a functionally important upstream negative regulator of _Sprr1a_ in the heart. KNOCKDOWN OF SPRR1A IN
MIR-150 KO MICE BLUNTS CARDIAC FIBROSIS POST-MI OBSERVED FOLLOWING MIR-150 DEFICIENCY To further determine the response of miR-150 KO;_Sprr1a__hypo/hypo_ mice to MI, we assessed the degree
of fibrosis using Masson’s trichrome staining and picrosirius red staining of the hearts at 8 weeks post-MI. We find larger regions of fibrosis in miR-150 KO hearts than in WT MI controls,
as reported previously [12]. We next observe reduced fibrosis post-MI in miR-150 KO;_Sprr1a__hypo/hypo_ hearts compared to miR-150 KO hearts (Figs. 4, 5A, B, and Supplementary Fig. 2).
MiR-150 KO MI hearts also exhibit increased expression of fibrotic _Col5a1_, _Col6a1_, _Col1a1_, _Col3a1_, and _Ctgf_ (Figs. 5C, D, and 6A–C) compared to expression in WT controls, but
miR-150 KO;_Sprr1a__hypo/hypo_ MI hearts exhibit decreased expression of these profibrotic genes (Figs. 5C, D, and 6A–C) compared to miR-150 KO controls. Next, our in vivo protein analysis
reveals significantly elevated levels of VIMENTIN and α-SMA in miR-150 KO MI mouse hearts compared to WT controls and significantly decreased levels of VIMENTIN and α-SMA in miR-150
KO;_Sprr1a__hypo/hypo_ hearts at 8 weeks post-MI compared to miR-150 KO controls (Fig. 6D, E, and Supplementary Fig. 3); this is consistent with the mRNA data for the profibrotic genes
(Figs. 5C, D, and 6A–C). Collectively, these results demonstrate for the first time that genetic knockdown of _Sprr1a_ significantly attenuates adverse postinfarct remodeling mediated by
miR-150 deletion. MIR-150 IN HUMAN CFS ELICITS PROTECTIVE EFFECTS IN PART THROUGH DIRECT FUNCTIONAL REPRESSION OF PROFIBROTIC SPRR1A Because of the cardiac upregulation of miR-150 by Carv
[11] concurrent with the downregulation of _Sprr1a_ [13], and the downregulation of miR-150 in CFs isolated from TAC mice [15] concurrent with the upregulation of _Sprr1a_ in CFs during MI
[13], we next studied primary adult human CFs (HCFs) to test whether miR-150 and _SPRR1A_ are inversely regulated in HCFs treated with Carv as well as HCFs subjected to H/R conditions.
Indeed, _SPRR1A_ is downregulated in HCFs subjected to H/R conditions after Carv treatment (Supplementary Fig. 4) concurrent with the upregulation of miR-150 [28]. We also observe that
_SPRR1A_ is increased in HCFs after H/R (Supplementary Fig. 4), consistent with our in vivo results in post-MI hearts and isolated CFs from ischemic myocardium [13]. Notably, we previously
reported that miR-150 is downregulated in HCFs after H/R [28]. Together with other previous reports on miR-150 downregulation in H/R and MI [12] as well as I/R [29, 30], our results indicate
that _Sprr1a_ is a critical functional target of miR-150 in CFs. Because _Sprr1a_ expression is upregulated in CFs isolated from ischemic mouse hearts [13] concurrent with the
downregulation of miR-150 in CFs isolated from TAC mice [15], and miR-150 negatively regulates mouse CF activation in vitro [15], we first confirmed whether a direct target of miR-150,
_SPRR1A_ is repressed by miR-150 in HCFs. Our loss-of-function studies indeed show that _SPRR1A_ is increased after miR-150 inhibition in HCFs (Fig. 7A, B). We next investigated whether
_SPRR1A_ regulates HCF activation. We first observe that _SPRR1A_ knockdown in HCFs decreases the expression of profibrotic _ACTA2_ and _CTGF_ (Fig. 7C and Supplementary Fig. 5), and miR-150
knockdown increases the expression of _ACTA2, CTGF_, and _POSTN_ (Supplementary Fig. 6). To further assess the effects of _SPRR1A_ knockdown, we examined HCF proliferation using
bromodeoxyuridine assay. We find that compared to controls, _SPRR1A_ knockdown decreased HCF proliferation (Fig. 7D–F) under both normoxic and H/R conditions. This is consistent with our
gene expression data, showing that HCFs with _SPRR1A_ knockdown have decreased mRNA levels of S-phase marker _PCNA_, mitosis (M) marker _AURKB_, and G2/M-phase marker CCNB1 compared with
controls (Supplementary Fig. 7). Moreover, our wound migration studies reveal that compared to controls, _SPRR1A_ knockdown decreased HCF migration (Fig. 8A–C) under both normoxic and H/R
conditions. This is consistent with our gene expression data, showing that _SPRR1A_ knockdown in HCFs subjected to H/R decreases mRNA levels of cell migration markers, _CTHRC1_ and _TNC_
compared with controls (Supplementary Fig. 8). _SPRR1A_ knockdown in HCFs also suppresses mRNA levels of CF differentiation markers, _COL4A1_, _COL8A1_, and _SRF_ (Supplementary Fig. 9), as
well as the protein levels of profibrotic α-SMA and FIBRONECTION (Supplementary Fig. 10). Because TGF-β1/SMAD signaling pathway plays a key role in CF activation, we next investigated the
role of SPRR1A in the regulation of TGF-β1 and SMADs. We observe that _SPRR1A_ knockdown in HCFs subjected to H/R decreases mRNA levels of _TGFB1_, _SMAD2_, and _SMAD3_ compared with
controls (Supplementary Fig. 11). This is consistent with our in vivo data, showing that _Sprr1a_ knockdown in mice decreases _Smad3_ expression as well as mRNA and protein levels of TGF-β1
compared with controls (Supplementary Figs. 12, 13). Our data thus suggest that SPRR1A is sufficient to increase HCF activation in part by activating TGF-β1/SMAD signaling pathway. Finally,
to establish the functional relationship between miR-150 and _SPRR1A_ in HCF activation, we applied an antimiR/siRNA-based rescue strategy to validate the functional relevance of the direct
miR-150 target _SPRR1A_. MiR-150 knockdown increases HCF proliferation (Fig. 7D–F and Supplementary Fig. 7) and migration (Fig. 8A–C and Supplementary Fig. 8), which are attenuated by siRNA
against _SPRR1A_ (Figs. 7D–F, 8A–C, Supplementary Figs. 7, 8). We also show that miR-150 knockdown increases the expression of profibrotic _TGFB1_, _SMAD2, SMAD3, COL1A1, COL3A1_, _COL4A1_,
_COL8A1_, and _SRF_ under normoxic and/or H/R conditions, which are attenuated by _SPRR1A_ knockdown (Supplementary Figs. 9, 11, 14). Taken together, our data indicate that profibrotic
SPRR1A is a key direct and functional target of miR-150 in HCFs and whole mouse hearts. DISCUSSION In this study, we identify the direct functional interaction between miR-150 and SPRR1A as
a new regulatory mechanism pertinent to both MI in mice and HCF activation. Mice deficient in miR-150 are sensitized to MI as indicated by the increased cardiac fibrosis, apoptosis,
inflammation, and damage as well as impairment of left ventricular function. Using a novel miR-150 KO;_Sprr1a__hypo/hypo_ mouse model, we demonstrate that _Sprr1a_ knockdown attenuates
excessive adverse postinfarct remodeling mediated by miR-150 loss. Of note, miR-150 KO;_Sprr1a__hypo/hypo_ rescues in part the phenotype exhibited in miR-150 KO, which reaches similar levels
shown in WT MI or higher levels than WT MI (Figs. 2–6 and Supplementary Figs. 1, 3). Using primary adult HCFs, we also discover for the first time that miR-150 functionally inhibits
profibrotic _SPRR1A_ such that the increased expression of _SPRR1A_ in HCFs lacking miR-150 results in a higher degree of sustained CF activation. Thus, the current study suggests that
SPRR1A is a crucial ischemic injury-responsive target of miR-150 in whole mouse hearts and HCFs and that miR-150 confers protective actions on both HF in mice and HCF activation by
repressing profibrotic SPRR1A. β1AR is predominantly expressed in the heart, and β-arrestin-mediated β1AR signaling elicits protective effects after Isoproterenol (ISO)-induced injury [31].
In our previous study, miR-150 was activated by the β-blocker Carv acting through β-arrestin-mediated β1AR protective signaling [11]. We recently reported that miR-150 is a critical
downstream mechanism by which β1AR-mediated β-arrestin signaling pathways confer protection [32]. Together with the results presented here, we posit that β-arrestin-mediated β1AR regulatory
mechanisms of miR-150 activation elicit beneficial remodeling in failing hearts by repressing CF activation through inhibiting profibrotic genes, including _Sprr1a_. Interestingly, we
previously showed that miR-150 deletion significantly compromised cardiac function and remodeling following MI by increasing cell death without affecting neovascularization [12], whereas Liu
Z et al. reported that miR-150 overexpression mediated by AgomiR injection protected the mouse heart against acute MI (AMI) by inhibiting monocyte migration [20]. More recently, using a
novel mouse model, we demonstrated that cardiac-specific miR-150 cKO mice had enhanced maladaptive post-MI remodeling. Mechanistically, miR-150 represses a proapoptotic and direct CM target,
_Sprr1a_ [13]. We also showed that _Sprr1a__hypo/hypo_ mice are protected against MI [13]. Although these previous studies showed the correlative relationship between miR-150 and _Sprr1a_
in HF, our overall knowledge of their functional actions remains elusive in part because of (i) the lack of mechanistic insight by which the miR-150/_Sprr1a_ dyad regulates HF and (ii) the
absence of rigorous studies to establish their direct in vivo functional relationship in HF. Our previous fractionation studies of different cardiac cell types showed that the expression of
miR-150 was significantly higher in normal CMs than CFs at baseline and its expression was not altered in stressed CMs, CFs, CIs, and cardiac endothelial cells (CEs) isolated from ischemic
myocardium at 1 week post-MI [13]. Other studies reported that cardiac-specific overexpression of miR-150 alleviates TAC-induced cardiac hypertrophy and HF [14] and that miR-150 loss results
in a higher degree of cardiac fibrosis after TAC and miR-150 is downregulated in CFs (not CMs) isolated from TAC mice [15]. MiR-150 also negatively regulates mouse CF activation in vitro
[15]. It is known that TGF-β1 or TGF-β/Smad signaling inhibits miR-150 expression [33, 34], coincident with SPRR1A upregulation [35]. MiR-150 can also suppress TGF-β in rat hearts and
primary CFs [36]. These previous studies indicate that miR-150 or SPRR1A plays an antifibrotic or profibrotic role, respectively. Interestingly, prior studies showed that SRF and c-Myb are
possible downstream mechanisms that explain antifibrotic effects of miR-150 in the heart [14, 15]. Liu W et al. reported that SRF is repressed by miR-150 in mouse hearts without presenting
any functional rescue experiments [14]. Deng P et al. showed that c-Myb is a key functional target of miR-150 in mouse CFs, but they did not establish their in vivo functional link [15].
Thus, the significance of SRF- and c-Myb-dependent mechanisms of miR-150 actions in vivo has not been defined. Here, we make novel discoveries to establish the functional miR-150/SPRR1A axis
in whole mouse hearts and HCFs as well as to define the role of SPRR1A in CF activation. SPRR1A is a known substrate of TGase II, and prior studies have linked TGase II to HF [37, 38] and
apoptosis of noncardiac cells [39, 40], suggesting a potential role of SPRR1A in HF and apoptosis. In agreement with this idea, we previously showed that CMs with knockdown of _Sprr1a_ are
protected against apoptosis in H/R and that _Sprr1a__hypo/hypo_ mice are protected against MI [13]. Interestingly, _Sprr1a_ expression is increased in post-MI hearts [13, 23] concurrent with
miR-150 downregulation [12]. Our previous cardiac cell fractionation study showed that _Sprr1a_ was ubiquitously expressed in CMs, CFs, CIs, and CEs at baseline. Despite this ubiquitous
expression pattern at baseline, _Sprr1a_ is upregulated in CMs isolated from mouse hearts post-MI [13], consistent with a report of SPRR1A upregulation in CMs from TAC-induced myocardium
[23]. Of importance, we also showed that _Sprr1a_ is upregulated in CFs isolated from post-MI hearts [13], suggesting a potential role of _Sprr1a_ in CFs and cardiac fibrosis. This notion is
supported by previous studies showing that adenovirus-mediated ectopic overexpression of _Sprr1a_ in vivo promotes cardiac fibrosis after TAC [23] and that SPRR1A levels are more highly
expressed in CFs from the infarct zone than in those from the remote region following MI [27]. Our current study also demonstrates for the first time that _SPRR1A_ knockdown suppresses HCF
proliferation and migration (Figs. 7, 8 and Supplementary Figs. 7, 8) and that _Sprr1a_ knockdown attenuates cardiac fibrosis post-MI observed following miR-150 deletion (Figs. 4–6 and
Supplementary Figs. 2,3). In our previous study, we also reported that LV _SPRR1A_ is upregulated in patients with HFrEF [13], consistent with other studies in mice with ISO-induced
myocardial injury [24] and renal I/R injury [25]. Notably, _SPRR1A_ expression was inversely associated with the survival of cancer patients [41,42,43]. Moreover, our recent study reported
that _Sprr1a_ is downregulated in hearts and CMs by Carv [13], concurrent with the upregulation of miR-150 [11]. Here, we also show that _SPRR1A_ is downregulated in HCFs by Carv
(Supplementary Fig. 4), concurrent with the upregulation of miR-150 [28]. Consistent with our findings, the cardiac upregulation of _Sprr1a_ after injury is suppressed by treatment with
cardioprotective Danshen [24]. Thus, the findings of _Sprr1a_ upregulation in CMs and CFs during MI further support that _Sprr1a_ inhibition could be therapeutically beneficial for HF and
cardiac fibrosis. Notably, a regeneration-associated gene, _Sprr1a_ was shown to be regulated by miR-463-3p in tibial nerve tissue [44] as well as by miR-155 in neurons and macrophages [45].
Except for these two other reports, little is known about the regulation of _Sprr1a_ by miRs. Given our findings that profibrotic SPRR1A is an important functional target of miR-150 in
whole mouse hearts (Figs. 1–6 and Supplementary Figs. 1–3) and HCFs (Figs. 7, 8, Supplementary Figs. 7–9, and Supplementary Figs. 11, 14) and that SPRR1A was regulated by miR-150 by direct
interaction [13], future targeted treatment options based on SPRR1A could be considered in MI patients with decreased levels of miR-150. LIMITATIONS Although we demonstrate that systemic
knockdown of _Sprr1a_ in mice alleviates maladaptive post-MI remodeling caused by miR-150 loss and that miR-150 is an important negative regulator of HCF activation in vitro by functionally
repressing profibrotic _SPRR1A_, miR-150 or _Sprr1a_ expression in other myocardial cells may also play a prominent role as supported by our recent study using CM-specific miR-150 cKO mice
[13]. Future studies using conditional cell-specific mouse models are thus warranted to fully understand the possible contribution of miR-150 or _Sprr1a_ expression in other cell types to
postischemic heart remodeling. Especially, whether _Sprr1a_ is a key downstream effector of fibroblast miR-150 during post-MI fibrotic remodeling in mice remains to be determined and is
beyond the scope of the current study. To expand our understanding of the sequence of events and to evaluate fibroblast roles in inflammatory and wound healing responses, additional
immunohistochemical assessments and gene expression studies (e.g., inflammation, acute cardiac cell death, and wound healing) during AMI are also needed. Notably, our functional rescue data
in mouse hearts and HCFs establish a functional link between miR-150 and SPRR1A in HF, supporting that they are in a linear pathway. Nevertheless, miR-150 may also have other targets
mediating distinct functions. We will investigate additional novel functional targets (i.e., SPRR1A-independent mechanisms) in the heart by cross-referencing the gene signature from
cardiac-specific miR-150 cKO mice [13] with miR-150 target prediction analyses in our future mechanistic studies. Importantly, the downstream targets and mechanisms of SPRR1A that regulate
CF activation remain elusive despite the suggestion in our current data (Supplementary Figs. 5 and 7–14) that SPRR1A activates profibrotic markers. Especially, a potential role of SPRR1A as
a regulator of the cytoskeleton remains to be determined because SPRR1A is localized selectively to actin-rich membrane ruffles and is colocalized with F-actin microfilaments [46]. Moreover,
additional methods, such as the Simpson method with multiple B-mode images and pressure-volume loop analysis would be required to measure cardiac function more accurately and to assess
diastolic dysfunction. Atomic force microscopy will be also needed to determine the stiffness of ventricular scar tissue post-MI. In the current study, we use a primary adult HCF model to
bolster the translational significance. Although this human cell model is an appropriate human cell source for HF remodeling in vitro, the mode of action in mice could be different in human
cells. We would employ CFs isolated from the mice used in this study to confirm the miR-150/SPRR1A axis in CFs. Although we show that modulating the miR-150/SPRR1A axis affects HCF
activation after H/R, most CFs are activated by recruited inflammatory cells after MI, not by ischemia. We would thus use TGF-β in future CF studies. Last, other in vivo injury models as
well as detailed studies on the other roles of the miR-150/SPRR1A dyad in all cell types are warranted before pursuing this axis as a vital therapeutic modality. CONCLUSIONS Our results
using a novel double loss-of-function mouse model and primary adult HCFs suggest that SPRR1A knockdown attenuates adverse post-MI remodeling mediated by miR-150 deletion and that miR-150
plays a vital protective role in part by blunting CF activation through its direct functional repression of profibrotic SPRR1A. Although miR-150 is associated with HF in humans [21] and the
correlative relationship between miR-150 and SPRR1A in the heart has been shown [13], our studies directly establish the functional relationship between miR-150 and SPRR1A during both
post-MI remodeling in mice and HCF activation as well as define an underlying mechanism by which miR-150 affects CF activation. Given that upregulation of SPRR1A [13, 23] or downregulation
of miR-150 [16, 29, 30, 47] also underlies other forms of cardiac disease, the deleterious action of SPRR1A and the protective action of miR-150 in whole mouse hearts and HCFs are likely
applicable to multiple stress settings. Therefore, reducing SPRR1A levels via SPRR1A knockdown and boosting miR-150 levels via Carv or miR-150 overexpression, in part to attenuate CF
activation, could be an attractive adjunctive strategy to provide therapeutic benefits. AVAILABILITY OF DATA AND MATERIALS All data are included in the manuscript and Supplementary
Information. The analytical methods and study materials will be made available to other researchers for the purposes of reproducing the results or replicating the procedures. Other methods
are provided in Supplementary Information. REFERENCES * Lei Z, Sluijter JP, van Mil A. MicroRNA therapeutics for cardiac regeneration. Mini Rev Med Chem. 2015;15:441–51. Article CAS PubMed
Google Scholar * Catalucci D, Gallo P, Condorelli G. MicroRNAs in cardiovascular biology and heart disease. Circ Cardiovasc Genet. 2009;2:402–8. Article CAS PubMed Google Scholar *
van Rooij E. The art of microRNA research. Circ Res. 2011;108:219–34. Article PubMed Google Scholar * Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, et al.
MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca(2)(+) overload and cell death. J Clin Investig. 2012;122:1222–32. Article CAS PubMed PubMed Central Google
Scholar * Arunachalam G, Upadhyay R, Ding H, Triggle CR. MicroRNA signature and cardiovascular dysfunction. J Cardiovasc Pharmacol. 2015;65:419–29. Article CAS PubMed Google Scholar *
Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–22. Article CAS PubMed Google Scholar *
Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee SS. Therapeutic miRNA and siRNA: Moving from bench to clinic as next generation medicine. Mol Ther Nucleic Acids. 2017;8:132–43. Article
CAS PubMed PubMed Central Google Scholar * Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J, et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice
weekly in patients with advanced solid tumors. Investig New Drugs. 2017;35:180–8. Article CAS Google Scholar * Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et
al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–94. Article CAS PubMed Google Scholar * Taubel J, Hauke W, Rump S, Viereck J, Batkai S, Poetzsch J, et
al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J.
2021;42:178–88. Article CAS PubMed Google Scholar * Kim IM, Wang Y, Park KM, Tang Y, Teoh JP, Vinson J, et al. Beta-arrestin1-biased beta1-adrenergic receptor signaling regulates
microRNA processing. Circ Res. 2014;114:833–44. Article CAS PubMed Google Scholar * Tang Y, Wang Y, Park KM, Hu Q, Teoh JP, Broskova Z, et al. MicroRNA-150 protects the mouse heart from
ischaemic injury by regulating cell death. Cardiovasc Res. 2015;106:387–97. Article CAS PubMed PubMed Central Google Scholar * Aonuma T, Moukette B, Kawaguchi S, Barupala NP, Sepulveda
MN, Corr C, et al. Cardiomyocyte microRNA-150 confers cardiac protection and directly represses proapoptotic small proline-rich protein 1A. JCI Insight. 2021;6:e150405. Article PubMed
PubMed Central Google Scholar * Liu W, Liu Y, Zhang Y, Zhu X, Zhang R, Guan L, et al. MicroRNA-150 protects against pressure overload-induced cardiac hypertrophy. J Cell Biochem.
2015;116:2166–76. Article CAS PubMed Google Scholar * Deng P, Chen L, Liu Z, Ye P, Wang S, Wu J, et al. MicroRNA-150 inhibits the activation of cardiac fbroblasts by regulating c-Myb.
Cell Physiol Biochem. 2016;38:2103–22. Article CAS PubMed Google Scholar * Devaux Y, Vausort M, McCann GP, Zangrando J, Kelly D, Razvi N, et al. MicroRNA-150: a novel marker of left
ventricular remodeling after acute myocardial infarction. Circ Cardiovasc Genet. 2013;6:290–8. Article CAS PubMed Google Scholar * Kreth S, Ledderose C, Schutz S, Beiras A, Heyn J, Weis
F, et al. MicroRNA-150 inhibits expression of adiponectin receptor 2 and is a potential therapeutic target in patients with chronic heart failure. J Heart Lung Transpl. 2014;33:252–60.
Article Google Scholar * Rhodes CJ, Wharton J, Boon RA, Roexe T, Tsang H, Wojciak-Stothard B, et al. Reduced microRNA-150 is associated with poor survival in pulmonary arterial
hypertension. Am J Respir Crit Care Med. 2013;187:294–302. Article CAS PubMed Google Scholar * Goren Y, Meiri E, Hogan C, Mitchell H, Lebanony D, Salman N, et al. Relation of reduced
expression of miR-150 in platelets to atrial fibrillation in patients with chronic systolic heart failure. Am J Cardiol. 2014;113:976–81. Article CAS PubMed Google Scholar * Liu Z, Ye P,
Wang S, Wu J, Sun Y, Zhang A, et al. MicroRNA-150 protects the heart from injury by inhibiting monocyte accumulation in a mouse model of acute myocardial infarction. Circ Cardiovasc Genet.
2015;8:11–20. Article CAS PubMed Google Scholar * Scrutinio D, Conserva F, Passantino A, Iacoviello M, Lagioia R, Gesualdo L. Circulating microRNA-150-5p as a novel biomarker for
advanced heart failure: a genome-wide prospective study. J Heart Lung Transplant. 2017;36:616–24. Article PubMed Google Scholar * Tesfaigzi J, Carlson DM. Expression, regulation, and
function of the SPR family of proteins. a review. Cell Biochem Biophys. 1999;30:243–65. Article CAS PubMed Google Scholar * Pradervand S, Yasukawa H, Muller OG, Kjekshus H, Nakamura T,
St Amand TR, et al. Small proline-rich protein 1A is a gp130 pathway- and stress-inducible cardioprotective protein. EMBO J. 2004;23:4517–25. Article CAS PubMed PubMed Central Google
Scholar * Wu P, Zhang Z, Ma G, Li J, Zhou W. Transcriptomics and metabolomics reveal the cardioprotective effect of Compound Danshen tablet on isoproterenol-induced myocardial injury in
high-fat-diet fed mice. J Ethnopharmacol. 2019;246:112210. Article PubMed Google Scholar * Su M, Hu X, Lin J, Zhang L, Sun W, Zhang J, et al. Identification of candidate genes involved in
renal ischemia/reperfusion injury. DNA Cell Biol. 2019;38:256–62. Article CAS PubMed PubMed Central Google Scholar * Reddy SP, Konkin T, Wu R. Structure and organization of the genes
encoding mouse small proline-rich proteins, mSPRR1A and 1B. Gene. 1998;224:59–66. Article CAS PubMed Google Scholar * Shah H, Hacker A, Langburt D, Dewar M, McFadden MJ, Zhang H, et al.
Myocardial infarction induces cardiac fibroblast transformation within injured and noninjured regions of the mouse heart. J Proteome Res. 2021;20:2867–81. Article CAS PubMed Google
Scholar * Aonuma T, Moukette B, Kawaguchi S, Barupala NP, Sepulveda MN, Frick K, et al. MiR-150 attenuates maladaptive cardiac remodeling mediated by long noncoding RNA MIAT and directly
represses profibrotic Hoxa4. Circ Heart Fail. 2022;15:e008686. Article CAS PubMed PubMed Central Google Scholar * Topkara VK, Mann DL. Role of microRNAs in cardiac remodeling and heart
failure. Cardiovasc Drugs Ther. 2011;25:171–82. Article CAS PubMed Google Scholar * Duan Y, Zhou B, Su H, Liu Y, Du C. MiR-150 regulates high glucose-induced cardiomyocyte hypertrophy by
targeting the transcriptional co-activator p300. Exp Cell Res. 2013;319:173–84. Article CAS PubMed Google Scholar * Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG,
Chen J, et al. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Investig. 2007;117:2445–58. Article CAS PubMed PubMed Central
Google Scholar * Moukette B, Kawaguchi S, Sepulveda MN, Hayasaka T, Aonuma T, Liangpunsakul S, et al. MiR-150 blunts cardiac dysfunction in mice with cardiomyocyte loss of
beta(1)-adrenergic receptor/beta-arrestin signaling and controls a unique transcriptome. Cell Death Discov. 2022;8:504. Article CAS PubMed PubMed Central Google Scholar * Davoodian P,
Ravanshad M, Hosseini SY, Khanizadeh S, Almasian M, Nejati Zadeh A, et al. Effect of TGF-beta/smad signaling pathway blocking on expression profiles of miR-335, miR-150, miR-194, miR-27a,
and miR-199a of hepatic stellate cells (HSCs). Gastroenterol Hepatol Bed Bench. 2017;10:112–7. PubMed PubMed Central Google Scholar * Zheng J, Lin Z, Dong P, Lu Z, Gao S, Chen X, et al.
Activation of hepatic stellate cells is suppressed by microRNA-150. Int J Mol Med. 2013;32:17–24. Article CAS PubMed Google Scholar * Zheng L, Zhou Z, Lin L, Alber S, Watkins S, Kaminski
N, et al. Carbon monoxide modulates alpha-smooth muscle actin and small proline rich-1a expression in fibrosis. Am J Respir Cell Mol Biol. 2009;41:85–92. Article CAS PubMed Google
Scholar * Gu Y, Zhang S, Chen X, Li Y, Liu Y. LongShengZhi alleviated cardiac remodeling via upregulation microRNA-150-5p with matrix metalloproteinase 14 as the target. J Ethnopharmacol.
2022;291:115156. Article CAS PubMed Google Scholar * Iwai N, Shimoike H, Kinoshita M. Genes up-regulated in hypertrophied ventricle. Biochem Biophys Res Commun. 1995;209:527–34. Article
CAS PubMed Google Scholar * Small K, Feng JF, Lorenz J, Donnelly ET, Yu A, Im MJ, et al. Cardiac specific overexpression of transglutaminase II (G(h)) results in a unique hypertrophy
phenotype independent of phospholipase C activation. J Biol Chem. 1999;274:21291–6. Article CAS PubMed Google Scholar * Piredda L, Amendola A, Colizzi V, Davies PJ, Farrace MG, Fraziano
M, et al. Lack of 'tissue' transglutaminase protein cross-linking leads to leakage of macromolecules from dying cells: relationship to development of autoimmunity in MRLIpr/Ipr
mice. Cell Death Differ. 1997;4:463–72. Article CAS PubMed Google Scholar * Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue
transglutaminase II. J Biol Chem. 2001;276:20673–8. Article CAS PubMed Google Scholar * Zhang H, Gao J, Zhao Z, Li M, Liu C. Clinical implications of SPRR1A expression in diffuse large
B-cell lymphomas: a prospective, observational study. BMC Cancer. 2014;14:333. Article PubMed PubMed Central Google Scholar * Chen G, Li G, Luo M, Wei X, Wang D, Zhang H, et al. Clinical
significance of SPRR1A expression in progesterone receptor-positive breast cancer. Tumour Biol. 2015;36:2601–5. Article CAS PubMed Google Scholar * Deng Y, Zheng X, Zhang Y, Xu M, Ye C,
Lin M, et al. High SPRR1A expression is associated with poor survival in patients with colon cancer. Oncol Lett. 2020;19:3417–24. CAS PubMed PubMed Central Google Scholar * Zhao J, Wu
C. MiR-463-3p inhibits tibial nerve regeneration via post-transcriptional suppression of SPRR1A artificial cells. Nanomed Biotechnol. 2019;47:3631–7. CAS Google Scholar * Gaudet AD,
Mandrekar-Colucci S, Hall JC, Sweet DR, Schmitt PJ, Xu X, et al. miR-155 deletion in mice overcomes neuron-intrinsic and neuron-extrinsic barriers to spinal cord repair. J Neurosci.
2016;36:8516–32. Article CAS PubMed PubMed Central Google Scholar * Bonilla IE, Tanabe K, Strittmatter SM. Small proline-rich repeat protein 1A is expressed by axotomized neurons and
promotes axonal outgrowth. J Neurosci. 2002;22:1303–15. Article CAS PubMed PubMed Central Google Scholar * Liu Z, Zhou C, Liu Y, Wang S, Ye P, Miao X, et al. The expression levels of
plasma micoRNAs in atrial fibrillation patients. PLoS ONE. 2012;7:e44906. Article CAS PubMed PubMed Central Google Scholar Download references FUNDING This work was supported by the
American Heart Association (AHA) Postdoctoral Fellowship 900453 to S.K., the AHA Career Development Award 931621 to M.N.S., the National Institutes of Health (NIH) R01HL148165 to S.J.C., and
the NIH R01HL146481 to I.K. AUTHOR INFORMATION Author notes * Satoshi Kawaguchi Present address: Department of Emergency Medicine, Asahikawa Medical University, Asahikawa, Hokkaido, Japan *
Bruno Moukette Present address: Internal Medicine Research Unit, Pfizer Inc., Cambridge, MA, USA * Tatsuya Aonuma Present address: Division of Cardiology, Nephrology, Pulmonology, and
Neurology, Department of Internal Medicine, Asahikawa Medical University, Asahikawa, Hokkaido, Japan * These authors contributed equally: Satoshi Kawaguchi, Bruno Moukette. AUTHORS AND
AFFILIATIONS * Department of Anatomy, Cell Biology, and Physiology, Indiana University School of Medicine, Indianapolis, IN, USA Satoshi Kawaguchi, Bruno Moukette, Marisa N. Sepúlveda, Taiki
Hayasaka, Tatsuya Aonuma, Angela K. Haskell, Jessica Mah & Il-man Kim * Division of Gastroenterology and Hepatology, Indiana University School of Medicine, Indianapolis, IN, USA Suthat
Liangpunsakul * Vascular Biology Center, Medical College of Georgia, Augusta University, Augusta, GA, USA Yaoliang Tang * Herman B Wells Center for Pediatric Research, Indiana University
School of Medicine, Indianapolis, IN, USA Simon J. Conway & Il-man Kim * Krannert Cardiovascular Research Center, Indiana University School of Medicine, Indianapolis, IN, USA Il-man Kim
Authors * Satoshi Kawaguchi View author publications You can also search for this author inPubMed Google Scholar * Bruno Moukette View author publications You can also search for this author
inPubMed Google Scholar * Marisa N. Sepúlveda View author publications You can also search for this author inPubMed Google Scholar * Taiki Hayasaka View author publications You can also
search for this author inPubMed Google Scholar * Tatsuya Aonuma View author publications You can also search for this author inPubMed Google Scholar * Angela K. Haskell View author
publications You can also search for this author inPubMed Google Scholar * Jessica Mah View author publications You can also search for this author inPubMed Google Scholar * Suthat
Liangpunsakul View author publications You can also search for this author inPubMed Google Scholar * Yaoliang Tang View author publications You can also search for this author inPubMed
Google Scholar * Simon J. Conway View author publications You can also search for this author inPubMed Google Scholar * Il-man Kim View author publications You can also search for this
author inPubMed Google Scholar CONTRIBUTIONS S.K., B.M., M.N.S., T.H., and I.K. designed the research studies, directed the study, and wrote the manuscript. S.K., B.M., M.N.S., and T.H.
conducted the experiments, acquired the data, analyzed the data, and prepared the figures. I.K. supervised the study and provided financial support. T.A., A.K.H., J.M., S.L., Y.T., and
S.J.C. helped to analyze the data and write the manuscript. S.K. and B.M. are listed as co-first authors. The order of co-first authors was determined mainly by the numbers of data presented
in this study, not via any other biases. CORRESPONDING AUTHOR Correspondence to Il-man Kim. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by Professor Sergio Lavandero.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY TEXTS, TABLES AND FIGURES REPRODUCIBILITY CHECKLIST FULL AND UNCROPPED WESTERN BLOTS RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under
a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article
are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kawaguchi, S., Moukette, B., Sepúlveda, M.N. _et al._ SPRR1A is a
key downstream effector of MiR-150 during both maladaptive cardiac remodeling in mice and human cardiac fibroblast activation. _Cell Death Dis_ 14, 446 (2023).
https://doi.org/10.1038/s41419-023-05982-y Download citation * Received: 02 March 2023 * Revised: 10 July 2023 * Accepted: 11 July 2023 * Published: 19 July 2023 * DOI:
https://doi.org/10.1038/s41419-023-05982-y SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative