Nf-κb-inducing kinase (nik) is activated in pancreatic β-cells but does not contribute to the development of diabetes
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The transcription factor nuclear factor-κB (NF-κB) has a key role in the pathogenesis of diabetes and its complications. Although activation of the canonical NF-κB pathway in
β-cells is generally deleterious, little is known about the role of the non-canonical NF-κB signalling and its main regulator, the NF-κB-inducing kinase (NIK), on pancreatic β-cell survival
and function. Previous studies based on models of NIK overexpression in pancreatic islet cells showed that NIK induced either spontaneous β-cell death due to islet inflammation or glucose
intolerance during diet-induced obesity (DIO) in mice. Therefore, NIK has been proposed as a potential target for diabetes therapy. However, no clear studies showed whether inhibition of NIK
improves diabetes development. Here we show that genetic silencing of NIK in pancreatic β-cells neither modifies diabetes incidence nor inflammatory responses in a mouse model of
immune-mediated diabetes. Moreover, NIK silencing in DIO mice did not influence body weight gain, nor glucose metabolism. In vitro studies corroborated the in vivo findings in terms of
β-cell survival, function, and downstream gene regulation. Taken together, our data suggest that NIK activation is dispensable for the development of diabetes. SIMILAR CONTENT BEING VIEWED
BY OTHERS NF-ΚB ACTIVITY DURING PANCREAS DEVELOPMENT REGULATES ADULT Β-CELL MASS BY MODULATING NEONATAL Β-CELL PROLIFERATION AND APOPTOSIS Article Open access 04 January 2021 IL-17F INDUCES
INFLAMMATION, DYSFUNCTION AND CELL DEATH IN MOUSE ISLETS Article Open access 04 August 2020 TBK1 REGULATES REGENERATION OF PANCREATIC Β-CELLS Article Open access 09 November 2020
INTRODUCTION Diabetes is one of the most prevalent chronic diseases worldwide, affecting more than 463 million people globally (9.3%). Projections show that if the rising trend of past
decades continues, 700 million (10.9%) people will be diabetic by 2045 [1]. Type 2 diabetes (T2D), which represents 90% of diabetes cases, is characterized by a systemic chronic low-grade
inflammation, insulin resistance and impaired function and survival of insulin-producing β-cells [2]. Type 1 diabetes (T1D), which accounts for around 10% of diabetic cases, is caused by
autoimmune-mediated destruction of β-cells, leading to severe hyperglycaemia [3]. β-cell death is a feature of both T1D and T2D, highlighting the crucial need for a better understanding of
this phenomenon and the development of interventions to preserve or restore β-cell mass. Activation of NF-κB is key in the pathogenesis of diabetes and its complications [4]. In T1D,
pro-inflammatory cytokines such as interleukin-1β (IL-1β), tumour necrosis factor (TNF) and CD40L, secreted by immune cells in the islets, induce the activation of NF-κB in β-cells via both
the canonical and non-canonical pathways [5, 6]. Although, in vitro and in vivo models of T1D have shown that activation of the canonical NF-κB pathway in β-cells is generally deleterious
[7], little is known regarding the role of the non-canonical NF-κB pathway in diabetes. The non-canonical NF-κB pathway is characterized by the recruitment of cellular inhibitors of
apoptosis 1 and 2 (cIAP1/2) by the TNF receptor-associated factor 2 (TRAF2) leading to TRAF3 proteolysis and accumulation of NIK. In turn, NIK phosphorylates IKKα leading to processing of
inhibitory protein p100 into the active subunit p52 that binds to RelB and translocates to the nucleus to induce gene expression [8, 9]. Some ligands involved in T1D progression, such as
lymphotoxin (LTα1β2), CD40L and the TNF superfamily 14 (TNFSF14, also named LIGHT) can activate the non-canonical NF-κB pathway [5, 8, 10, 11]. We have previously observed in vitro that
knocking down of p100 decreased cytokine-mediated apoptosis and inflammatory responses in rodent β-cells [12], indicating a role for the non-canonical NF-κB pathway in β-cell demise.
Recently, two studies showed that NIK overexpression in β-cells resulted in impaired glucose-stimulated insulin secretion (GSIS), however diverged regarding effects on islet inflammation and
β-cell survival [13, 14]. While, transgenic overexpression of NIK specifically in β-cells (β-NIK-OE mice) resulted in early spontaneous diabetes onset in mice due to insulitis and β-cell
death, mice expressing NIK constitutively in β-cells due to TRAF2/3 depletion showed no diabetic phenotype up to 16 weeks of age with a mild glucose intolerance under control chow diet [13,
14]. It is important to consider that sustained NIK overexpression is not representative of physiological conditions because NIK is constantly degraded [15, 16], and even in optimal
stimulatory conditions its level is generally low. Thus, NIK overexpression is not optimal to study the effects of this kinase. To better understand the role of NIK and the non-canonical
NF-κB in immune-mediated β-cell death we developed a β-cell-specific NIK knockout (_NIKβ_KO) mice. Under physiological conditions lack of NIK did not affect β-cell development or function
and glucose homoeostasis. After multiple low-dose streptozotocin (MLDSTZ) treatment, metabolic parameters including glycemia, β-cell mass and recruitment of immune-cells to the islets were
indistinguishable between _NIKβ_KO and wild type (WT) littermates. Additionally, body weight gain and glucose metabolism were not different in _NIKβ_KO mice as compared to their WT
littermates after diet-induced obesity (DIO). Finally, we showed that specific ligands of the non-canonical NF-κB pathway did not affect β-cell death, cytokine/chemokine expression and
insulin secretory function in mouse islets or human β-cells. Taken together, our data suggests that although NF-κB activation is involved in diabetes pathogenesis, the non-canonical NF-κB
pathway led by NIK activation is dispensable for the development of diabetes in mice. RESULTS NIK IS NOT NECESSARY FOR PANCREATIC Β-CELL DEVELOPMENT AND THE ABSENCE OF NIK DOES NOT ALTER
Β-CELL FUNCTION AND GLUCOSE HOMOEOSTASIS IN MICE To assess the in vivo role of endogenous NIK expression in β cells, we generated _NIKβ_KO by using NIKfl/fl mice (gift from E. Dejardin) and
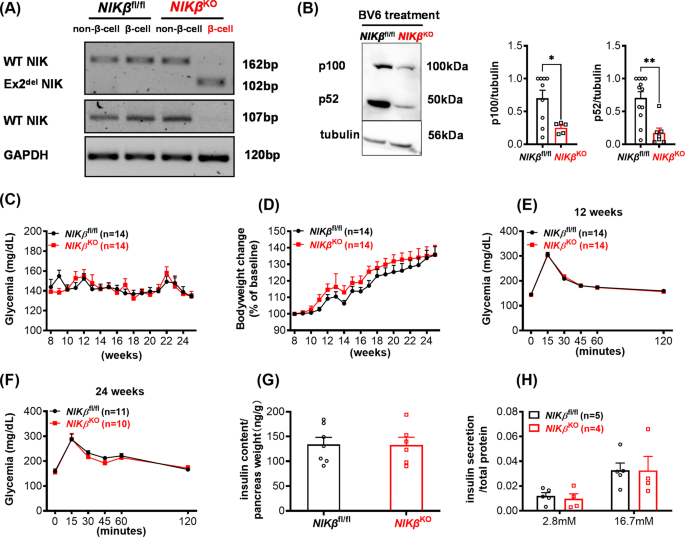
crossing with RIP-CRE mice [17]. mRNA analysis of FACS purified β-cells from _NIKβ_KO mice confirmed that they expressed a mutated NIK mRNA, while non-mutated NIK mRNA was detected in the
non-β-cell populations from _NIKβ_KO mice and both β-cell and non-β-cell populations from the WT mice (Fig. 1A). As shown in Fig. 1B, _NIKβ_KO mouse islets had decreased expression of both
p100 and p52 proteins as compared to WT mice, when treated with the second mitochondrial-derived activator of caspase (SMAC) mimetic BV6, which inhibits cIAPs leading to NIK stabilization
[18]. This is expected since p100 is positively regulated by NIK [19, 20] and thereby we confirmed that _NIKβ_KO mice had disrupted NIK signalling. Male and female _NIKβ_KO mice were
monitored weekly for fed blood glucose and bodyweight and no difference was found between _NIKβ_KO mice and WT littermates (Figs. 1C, D, S1A, B). At 12 and 24 weeks of age, NIK deletion did
not affect glucose tolerance between genotypes (Figs. 1E, F, S1C). At 24 weeks of age, no differences in total pancreatic insulin content were detected between _NIKβ_KO and WT mice (Fig.
1G). Finally, β-cell function was evaluated by performing GSIS and NIK depleted β-cells showed normal insulin secretory responses (Fig. 1H). Overall, our data suggest that _NIKβ_KO mice have
healthy β-cells and normal glucose homoeostasis under physiological conditions. NIK IS DISPENSABLE FOR THE DEVELOPMENT OF IMMUNE-MEDIATED DIABETES IN MICE To verify if NIK activation played
a role in immune-mediated β-cell death and diabetes development in vivo_, NIKβ_KO and WT mice were administered MLDSTZ treatment. The low doses of STZ are specifically toxic to β-cells
generating localized inflammation which is comparable to the inflammatory process described in human pancreas during T1D and the autoimmune nonobese diabetic (NOD) mouse model, namely,
insulitis with initial attraction of neutrophils and macrophages followed by T cells, which causes progressive decrease in insulin levels due to β-cell destruction [21,22,23,24]. Both
_NIKβ_KO and WT mice developed hyperglycaemia at 7 days after last STZ injection (Fig. 2A). At the end of follow up, as expected, mice injected with MLDSTZ had impaired glucose tolerance as
compared to buffer mice, although no difference between _NIKβ_KO mice and WT littermates was observed (Fig. 2B). MLDSTZ treated _NIKβ_KO and WT mice showed similar body weight during the
experiment (Fig. 2C). The mice were sacrificed at 45 days after the last injection of STZ. At this time point, MLDSTZ-treated mice showed low levels of pancreatic insulin content, residual
β-cell mass and islet density, while the difference was indistinguishable between genotypes (Fig. 2D–G). No difference was observed in the insulitis score between WT and _NIKβ_KO mice (Fig.
S2). A more refined analysis of immune responses was performed in MLDSTZ-treated mice sacrificed around 2 weeks after the last STZ injection, when full blow insulitis has been observed [23].
At this time point, mice are becoming hyperglycaemic and are glucose intolerant (Fig. 3A–C). However, no differences in these parameters were noticed between _NIKβ_KO and WT mice. We
observed that MLDSTZ significantly increased the frequency of regulatory T cells (Tregs, CD4+Foxp3+) in pancreatic draining lymph nodes (pLN) compared to buffer controls, however no
difference between _NIKβ_KO and WT littermates were detected (Fig. 3D). In the spleen and blood, again increased frequency of Tregs were found in MLDSTZ mice but were similar between
_NIKβ_KO mice and WT littermates (Fig. S3A). Next, the frequencies of IFN-γ+Th1 (CD4+) and cytotoxic (CD8+) T cells were analysed, we found that Th1 were also increased in pLn of
MLDSTZ-treated mice (although not statistically significance), cytotoxic T cells were not altered by MLDSTZ, and NIK absence did not affect frequency of IFN-γ+Th1 nor cytotoxic T cells (Fig.
3E, F). In the spleen, Th1 and cytotoxic T cells tended to be increased in MLDSTZ mice compared to controls, however no differences in blood or between _NIKβ_KO and WT mice were found (Fig.
S3B, C). We also analysed the frequencies of CD4+ and CD8+ effector/memory (CD44+CD62L−) T cells in pLn of mice and CD4+ effector memory T cells tended to be higher in MLDSTZ mice (although
not statistically significant) (Fig. 3G, H). In blood, both CD4+ and CD8+ effector/memory cells were increased in MLDSTZ mice, no changes were found in spleen or between _NIKβ_KO and WT
mice (Fig. S3E, F). Interestingly, we found a significant increase of a rare population of double-positive CD4+CD8+ T cells linked to autoimmunity and chronic inflammatory diseases [25] in
pLn of both _NIKβ_KO and WT MLDSTZ-treated mice (Fig. 3I). Taken together, these results suggest that NIK absence in β-cell does not change in vivo glycaemic response nor inflammatory
responses to islets in homoeostasis or MDLSTZ-induced diabetes. NIK IN Β-CELLS DOES NOT AFFECT GLUCOSE TOLERANCE NOR INSULIN RESISTANCE IN DIET-INDUCED OBESITY (DIO) Next, we assessed the
role of NIK in β-cells in DIO. During the 12 weeks of DIO, _NIKβ_KO and WT littermates presented normal blood glucose (Fig. 4A) and the two mouse strains gained weight similarly (Fig. 4B).
The nuclear magnetic resonance (NMR) analysis showed that after 12 weeks of HFD both _NIKβ_KO and WT littermates had a significant increase in their fat mass, while a reduction in lean mass
was observed. However, they showed equivalent percentages of fat mass and lean mass before and after DIO (Fig. 4C). Following the challenge with glucose, HFD-treated mice were glucose
intolerant, but no difference was found between the two genotypes (Fig. 4D). Moreover, no different responses were observed at the level of insulin tolerance tests between _NIKβ_KO and WT
mice (Fig. 4E). These results demonstrate that NIK expression in β-cells does not influence the adverse metabolic consequences of DIO. NIK ABSENCE DOES NOT MODIFY Β-CELL DEATH NEITHER
AFFECTS INFLAMMATORY GENE EXPRESSION We then further investigated the role of NIK on β-cell viability and inflammatory responses in islets from _NIKβ_KO and WT mice and in the human β-cell
line (EndoC-βH1 cells) [26, 27]. Treatment of mouse islets with ligands of the alternative NF-κB pathway, namely, lymphotoxin beta receptor agonist (LTβRa) [10, 28] or LIGHT did not induce
islet cell death and showed no additive effect on the cell death mediated by IL-1β+IFN-γ. Moreover, islets from _NIKβ_KO showed the same sensitivity to cell death as WT islets (Fig. 5A).
Streptozotocin exposure led to the significant death of mouse islets cells in a dose-dependent manner, while no difference between _NIKβ_KO and WT mouse islets was shown (Fig. 5B). In the
human β-cell line, the non-canonical NF-κB pathway was activated when treated with LIGHT, LTβRa (Fig. S4A–D). However, these specific ligands did not induce β-cell death (Fig. 5C). To verify
the effect of NIK in human β-cell survival we knocked down NIK using siRNA (Figs. 5D, S4A, S4,C, S4,E, S4,G) and treated the cells with IL-1β+IFN-γ. IL-1β+IFN-γ-induced NIK stabilization
and increased both the expression of p100 and p52 (Fig. S4G–H). However, NIK knockdown did not modify cytokine-mediated death of EndoC-βH1 cells or human islets (Fig. 5E, F). We then
compared how cytokines or NIK-specific ligands regulated NF-κB-dependent genes expression in human β-cells. Based on time course analysis, IL-1β+IFN-γ exposure significantly upregulated
NF-κB-dependent genes Fas, Ccl2, Cxcl1, and Cxcl10 whereas LIGHT and LTβRa had no effect on the expression of these genes (Fig. 5G). To confirm that NIK did not affect Fas nor chemokine
expression, we knocked down NIK in EndoC-βH1 cells exposed to IL-1β+IFN-γ or LIGHT for 16 h. As observed in Fig. 5H, expressions of Fas, Ccl2, Cxcl1, Cxcl10 were not modified by NIK
silencing. A similar effect was observed in mouse islets, in which neither LIGHT nor NIK absence had significant effect on gene expression, except for Cxcl10 that was decreased in islets
from _NIKβ_KO in response to exposure of IL-1β+IFN-γ (Fig. 5I). Overall, these data indicate that NIK absence has no major role in pancreatic β-cell death nor proinflammatory transcriptional
regulation. ACTIVATION OF NON-CANONICAL NF-ΚB DOES NOT AFFECT INSULIN SECRETORY FUNCTION IN MOUSE PANCREATIC ISLETS AND HUMAN Β CELLS NIK overexpression has previously been shown to inhibit
GSIS in β-cells both in vitro and in vivo [13, 14]. Moreover, LIGHT has also been shown to inhibit GSIS in human islets [10]. However, in our study, exposure of WT or _NIKβ_KO mouse islets
to LIGHT had no effect on GSIS (Fig. 6A). The mRNA expression of insulin was also not regulated by LIGHT or NIK in mouse islets (Fig. 6B). In line with that, EndoC-βH1 cells did not show
impaired insulin secretion when exposed to LTβRa and BV6 (Fig. 6C). Of note, BV6 induced NIK activation in EndoC-βH1 cells (Fig. S4E, F). Furthermore, exposure to LIGHT did not affect
insulin mRNA expression in the presence or absence of NIK (Fig. 6D). These data go against a “physiological” effect of NIK on β-cell function. DISCUSSION NIK is the key kinase of
non-canonical NF-κB signalling pathway, and its dysregulated expression has been found to play a role in many autoimmune diseases, such as systemic lupus erythematosus and rheumatoid
arthritis [29, 30]. Contrary to the well-known involvement of the canonical NF-κB signalling in T1D and T2D islet inflammation [6, 31], the role of NIK and the non-canonical NF-κB signalling
in diabetes pathology is unclear. Genome-wide association studies have identified a single-nucleotide polymorphism SNP rs17759555 of MAP3K14/NIK as a susceptibility gene of T1D [32] and
recent studies using NIK overexpression in mice have shown negative effects of NIK on β-cell survival and function in models of diabetes. Importantly, NIK overexpression is not a
physiological phenomenon and although NIK accumulation can temporarily occur under specific conditions to activate the non-canonical pathway, its expression is generally low due to
constitutive ubiquitin-mediated protein degradation [8]. To overcome this issue, we developed a NIK floxed mice and produced a β-cell specific NIK KO mice (_NIKβ_KO), which enabled us to
study the role of physiological NIK expression in diabetes development. Contrary to previous studies, _NIKβ_KO mice did not show any abnormality in their glucose metabolism under
physiological conditions, indicating that NIK is not necessary for embryonic development of β-cells and lack of NIK does not affect β-cell function [33]. To verify a possible role for NIK in
immune-mediated β-cell death we exposed the mice to MLDSTZ to induce immune-mediated diabetes. We did not observe any differences in the incidence nor timing of diabetes development between
_NIKβ_KO and wild type mice. Moreover, the glycaemia levels of both _NIKβ_KO and wild type littermates were similar. In agreement with these data, insulin content, β-cell mass and islet
area were not affected by NIK absence in this model. A previous study has shown that administration of a chemical inhibitor of NIK, B220, to high-dose STZ-treated mice improved the
hyperglycaemia, glucose intolerance and even restored β-cell mass [13]. These results are surprising since we did not observe any protection in vitro when exposing _NIKβ_KO mouse islets to
different doses of STZ, neither did we observe a protection in mice treated with MLDSTZ. Of note, high dose STZ is not a model of inflammation mediated β-cell apoptosis, since it induces a
fast and massive β-cell death mostly via necrosis [34, 35]. Another point to consider is that inhibitors often have non-specific targets. In line with this, a recent publication using a
considered highly selective NIK inhibitor, named, SMI affects the activity of at least three other kinases, namely mitogen-activated protein kinase kinase kinase kinase 5 (MAP4K5),
leucine-rich repeat kinase 2 (LRRK2), and protein kinase D1 (PKD1, PKCμ) [29, 36]. Thus, the use of chemical inhibitors of NIK which are less specific may lead to broader effects than
observed by the outcome of specific genetic knockout models, such as our _NIKβ_KO. In T1D, uncontrolled immune responses in the pancreas, particularly mediated by autoreactive T cells play a
significant role in β-cell death [37]. Therefore, we compared different T cell subtypes in buffer and diabetic _NIKβ_KO and _NIKβ_fl/fl mice, at 2 weeks after last STZ injection, when T
cells are described to be significantly increased in pLn of MLDSTZ-diabetic mice [38, 39]. In our study, MLDSTZ induced higher frequencies of CD4 effector T cells (IFN+, CD44+CD62L−), Tregs
(CD4+Foxp3+) in pLn of diabetic mice, indicating as expected that MLDSTZ provoked significant inflammation in the pancreas. The accumulation of Tregs in inflamed sites, especially in the
draining lymph nodes is conducive to optimal suppression of antigen-specific T effector responses [40, 41]. Moreover, a rare population of double positive (DP) CD4+CD8+T cells was also
significantly increased in pLN of diabetic MLDSTZ mice, the DP T cells are present in healthy individuals but have been shown to be increased in several pathologies such as infections,
neoplasias and some autoimmune diseases. They are described as having an effector/memory phenotype with enhanced cytolytic capacity [25, 42]. To our knowledge this is the first time that DP
have been described in an autoimmune diabetic model. It turned out, however, that NIK deletion in β-cells did not elicit any differences in the T cell responses in either the local pLn or
peripheral systems such as spleen and blood. Our results are significantly different from the results in β-NIK-OE mice, in which large infiltration of T cells correlated with extensive
β-cell loss. This is probably due to the fact that forced NIK overexpression triggers both the canonical and non-canonical NF-κB activation [43,44,45]. Our in vitro data showed that NIK
activation via specific ligands of the non-canonical NF-κB pathway did not induce death of human β-cells and islet cells. Moreover, inhibition of NIK was unable to prevent death of human
β-cells and mouse islets induced by different conditions, such as IL-1β + IFN-γ or STZ. Additionally, we also observed in both human β-cells and mouse islets that the specific non-canonical
ligands and NIK activation do not result in activation of Fas and several chemokines analysed indicating that the activation of these genes are occurring mostly via the canonical NF-κB
pathway [13, 43, 46]. The only exception was Cxcl10, its expression seems to be modulated by NIK in mouse β-cells but not in the EndoC-βH1 cells. The differences observed in the two cellular
models are probably due to species differences. It should also be considered that the mouse β-cells are primary non-dividing cells and the EndoC-βH1 is a cell line. Although expression of
CXCL10 can be involved in insulitis [47,48,49], we did not observe any impact of the absence of NIK in our MLDSTZ model. This may indicate that absence of β-cell-mediated expression of
CXCL10 is not enough to prevent insulitis or that in vivo CXCL10 expression was not modulated by NIK in β-cells. Overall, the in vitro data, reinforce the results obtained by the in vivo
studies showing a disconnection between NIK activation and β-cell-mediated inflammation and β-cell death. NIK overexpression due to TRAF2/3 depletion led to impaired insulin secretion in DIO
mice, mostly via inhibition of GSIS glucose [10, 13, 14]. Our data in _NIKβ_KO do not confirm any effects of NIK on glucose tolerance nor insulin resistance in DIO. Additionally, our
present findings do not support a role for NIK in the regulation of β-cell function as we did not observe inhibition of GSIS neither modification of insulin mRNA expression in β-cells in
conditions of NIK activation. Likewise, NIK absence/deletion in both mouse islets and human β-cells did not modify insulin mRNA expression. The different results between our studies and the
previous one is that while in our study NIK is activated via its endogenous ligands, NIK overexpression can produce non-specific responses as discussed above. Overall, our data suggests that
ablation of NIK has no major effects in β-cells both in vitro and in vivo. Therefore, we postulate that NIK and the non-canonical NF-κB pathway do not play a significant role in β-cell
insulitis and diabetes development. MATERIALS AND METHODS MATERIALS The cytokine concentrations utilized were based on prior studies [12, 50, 51] and are described in Supplementary Table 1.
For NIK detection by western blots EndoC-βH1 cell were treated with the proteasome inhibitor MG-132 (Sigma-Aldrich, Diegem, Belgium) at 10 µmol/L for the last 8 h before being harvested.
CULTURE AND TRANSFECTION OF ENDOC-ΒH1 AND DISPERSED HUMAN ISLETS CELLS EndoC-βH1 cells were purchased from UNIVERCELL-BIOSOLUTIONS (MTA BH1-201601171) and cultured in low-glucose DMEM
supplemented with 2% BSA fraction V, β-mercaptoethanol 50 µM, l-glutamine 1%, penicillin/streptomycin 2%, nicotinamide 10 mM, human transferrin 5.5 µg/mL and sodium selenite 6.7 ng/mL (all
from Sigma Aldrich, Diegem, Belgium) [52]. The dispersed human islets from human organ donors were prepared as previously done [53]. Small interfering (si)RNAs (30 nmol/L) used are listed in
Supplementary Table 2 and transfections were performed using lipofectamine RNAimax (Fisher Scientific, Aalst, Belgium) as described [50, 54, 55]. GENERATION AND CHARACTERISATION OF A
Β-CELL-SPECIFIC NIK KNOCKOUT MOUSE STRAIN, ISLET ISOLATION, Β-CELL SORTING AND CELL CULTURE NIKfl/fl (gift from Prof. Dejardin, GIGA, University of Liege, Liege, Belgium) were crossed with
RIP-Cre transgenic mice [56] to generate β-cell-specific NIK knockout mice _NIKβ_KO. Both lines are on the C57BL/6 genetic background and WT littermates were used as controls. _NIKβ_KO mice
were born at the expected normal Mendelian ratio. The non-fasted-glycaemia and body weight were followed in male and female _NIKβ_KO mice and their respective WT littermates from 8 to 24
weeks. An intra-peritoneal glucose tolerance test (ipGTT) was performed in these animals at 12 and 24 weeks of age. Mice were injected with 2 g/kg body weight glucose after 6 h of fasting.
At 24 weeks, mice were sacrificed, and their pancreas collected for measuring the total pancreatic insulin content [57]. For islet isolation, mouse pancreases were digested by collagenase
and incubated in a water bath at 37 °C. The islets were separated by a density gradient (Histopaque-1077; Sigma Aldrich), and then handpicked under a stereomicroscope [58]. These islets were
cultured and treated as described [54]. For FACS purification, single mouse islet cell preparations were obtained, and the sorting of a β-cell-enriched cell populations was performed in a
FACSAria instrument (BD Bioscience, San Jose, CA, USA) as described [59, 60]. RT-PCR using specific primers (Supplementary Table 4) designed for detecting exon 2 deletion of NIK were
performed on FACS purified pancreatic β- and non-β-cells from both _NIKβ_KO mice and WT littermates. Forward primer 1 is located at exon1 and forward primer 2 is at exon 2 which is flanked
by loxP sites and deleted in _NIKβ_KO mice, the common reverse primer is located at exon 3. GAPDH was used as loading control. Glucose-stimulated insulin secretion (GSIS) was performed in
freshly isolated islets [57, 61]. Insulin was quantified using the Ultra-Sensitive Mouse Insulin ELISA Kit (Crystal Chem, Downers Grove, USA). The GSIS experiments were performed and
measured in triplicates. MULTIPLE LOW-DOSE STREPTOZOTOCIN TREATMENT Non-fasted male mice aged 7–8 weeks were randomly divided to be injected i.p. for 5 consecutive days with either 42.5
mg/kg body weight streptozotocin (Sigma-Aldrich, Belgium) dissolved in citrate buffer (100 mM pH ≤ 4.5, made freshly) or citrate buffer alone. Blood glucose levels were measured on days −3,
1, 2, 3, 4, 5 pre- and post-injection and later weekly during 7 weeks (for long term analysis) or 2 weeks (for short term analysis) after the last injection, in non-fasting conditions, using
a glucometer (Accu-Chek, Roche, Switzerland) [62]. Hyperglycaemia was defined as non-fasting blood glucose levels >200 mg/dL in two sequential measurements. At the end of the experiment,
the animals were sacrificed, and the pancreas collected for histological analysis or for measuring the insulin content. DIET-INDUCED OBESITY MODEL (DIO) Male mice aged 8–9 weeks were
randomly selected to be fed a high fat diet (HFD) containing 40 kcal% fat (mostly palm oil), 20 kcal% fructose and 2% cholesterol (D09100310i, Research Diets, New Brunswick, NJ) for 12
weeks. Glycemia and bodyweight were monitored weekly. Lean and fat mass were analysed using EchoMRI™ 3-in-1 (NMR) body composition analyzer (EchoMedical Systems, Houston, TX) at weeks 8
(prior to start of the diet) and 20 (12 weeks of the diet). Mice were fasted for 6 h for the ipGTT and 4 h for the insulin tolerance test (iTT). FACS ANALYSIS OF IMMUNE CELLS Single cell
suspensions of pancreatic lymph nodes, spleen and blood were prepared from mice 14 days after last day of streptozotocin or buffer injections. The following antibodies were used for surface
staining: CD45 (cat. 56-0451-82), CD3e (cat. 45-0031-82), CD8 (cat. 63-0081-80), CD44 (cat. 25-0441-81), CD62L (cat. 11-0621-81), CD11b (cat. 63-0112-80), Ly-6G/C (cat. 47-5971-80), F4/80
(cat. 45-4801-80), CD86 (cat. 25-0862-80), (all from eBioscience, San Diego, USA), CD4 (cat. A15384) (from Invitrogen, Merelbeke, Belgium) Intracelular mAb against IFN-γ (cat. 12-7311-81),
FoxP3 (cat. 17-5773-82) were from eBioscience and used according to the manufacturer’s instructions. For viability staining, Zombie Violet™ Fixable Viability Kit (cat. 423113, Biolegend, San
Diego, USA) was used. Data were acquired using a BD LSR Fortessa™ X-20 Cell Analyzer (BD) instrument running FACS DIVA software and were analysed using FlowJo v10 (TreeStar, Ashland, OR).
Investigators analysing FACS data were blinded to mouse genotype and treatment groups. HISTOLOGY OF PANCREAS, BETA CELL FRACTIONAL AREA AND INSULITIS SCORE ANALYSIS Pancreata from sacrificed
mice were collected and included in paraffin. To evaluate the beta-cell fractional area, formalin-fixed paraffin embedded (FFPE) tissue sections (5-μm thickness) were prepared by using a
microtome (cat. RM2125 RTS-Leica Microsystems, Wetzlar, Germany) and baked overnight at 37 °C. After deparaffinization and rehydration through decreasing alcohol series (Xylene-I 20 min,
Xylene-II 20 min, EtOH 100% 5 min, EtOH 95% 5 min, EtOH 80% 5 min, EtOH 75% 5 min) pancreatic tissue sections were incubated with 1× phosphate-buffered saline with Ca2+ and Mg2+ (PBS 1×)
supplemented with 3% H2O2 (cat. H1009-Sigma Aldrich, St. Louis, MO, USA) for 40 min to block endogenous peroxidases. Heat-induced antigen retrieval was performed using 10 mM citrate buffer
pH 6.0 in microwave (600 W) for 10 min, maintaining boiling conditions. Sections were incubated with PBS 1× supplemented with 3% bovine serum albumin (BSA, cat. A1470-25G, Sigma Aldrich, St.
Louis, MO, USA) to reduce antibodies non-specific binding. Then, sections were incubated with primary antibody polyclonal Guinea Pig anti-Insulin (diluted 1:5 in 3% BSA, cat. IR002, Agilent
Technologies, Santa Clara, CA, USA) for 1 h at RT. Subsequently, sections were incubated with secondary antibody Goat anti-Guinea Pig HRP-conjugate (cat. 106-036-003), Jackson
ImmunoResearch, Philadelphia, PA, USA), diluted 1:2000 in PBS 1× for 1 h at room temperature (RT). Sections were then incubated with one drop of 3,3′-diaminobenzidine (DAB) chromogen
solution (cat. RE7270-K, Novolink MAX DAB, Leica Microsystems, Wetzlar, Germany) for ~2 min to trigger the chromatic reaction. Stained sections were then counterstained with hematoxylin
(cat. MHS31, Sigma Aldrich, St. Louis, MO, USA) for 4 min for better visualization of the tissue morphology. After the dehydration through increasing alcohol series, the pancreatic sections
were mounted with Eukitt mounting medium (cat. S9-25-37, Bio Optica, Milan, Italy) and covered with a coverslip allowing them to dry. Images were acquired using optical microscope (cat. M570
E, Eclipse Ni-U, Nikon, Tokyo, Japan). For each section, islets were acquired at ×20 magnification and total section area were measured using NIS-elements viewer software analysis (vs.
4.40.00). For each islet, insulin positive area was evaluated using ImageJ software (vs. 1.8.0). The sum of each insulin positive islet area was normalized to the area of the whole section
(reported as mm2) in order to obtain the β-cell fractional area. The islet density value was calculated by counting the total number of islets in the section and normalizing that with the
area of the whole section (reported as mm2). Insulitis score was assessed as previously done [63], by assigning a score of the islet infiltration to each islet analysed, as follows: 0, no
infiltration; 1, peri-insulitis; 2, islets with <50% of infiltration; 3, islets with >50% of infiltration. Investigators performing histological analysis were blinded to mouse genotype
and treatment groups. QUANTITATIVE RT-PCR AND WESTERN BLOT ANALYSIS Poly(A)+mRNA was isolated and reverse-transcribed as described [54]. The real-time PCR amplification reaction was
performed using SYBR Green and compared with a standard curve [64]. Expression values were corrected for the housekeeping gene GAPDH. All primers used are listed in Supplementary Table 3.
For Western blot analysis, cells and islets were washed once with cold PBS and then lysed with RIPA buffer supplemented with proteinase cocktail inhibitor [65]. Denatured lysates were then
resolved by SDS–PAGE and transferred to a nitrocellulose membrane. Western blot analysis was performed as described [65]. The following antibodies were utilized: anti-human NF-κB2 antibody
(cat. 05-361, Merck KGaA, Darmstadt, Germany) [66]; anti-mouse NF-κB2 antibody (cat. 4882S, Cell Signaling technology, Leiden, The Netherlands), anti-NIK antibody (cat. 4994S, Cell Signaling
technology, Leiden, The Netherlands) [12]; anti-GAPDH human polyclonal antibody (cat. 2275-PC-100, Trevigen, Gaithersburg, USA); polyclonal anti-tubulin (cat. T9026, Sigma-Aldrich Diegem,
Belgium), and horseradish peroxidase-conjugated goat anti-rabbit (cat. P044801-2) or anti-mouse (cat. P044701-2) IgG from Agilent (Santa Clara, United States) [12, 58, 60]. ASSESSMENT OF
CELL VIABILITY The percentages of viable cells were determined using the DNA-binding dyes propidium iodide (PI, 5 µg/mL, Sigma-Aldrich) and Hoechst 33342 (HO, 5 µg/mL, Sigma-Aldrich), as
described [65]. For mouse islets, the percentages of dead cells were evaluated in a minimum of 10 islets per condition. All assessments were performed by two independent researchers one of
whom was unaware of the identity of the samples. STATISTICAL ANALYSIS Data are presented as means ± SEM and were analysed using GraphPad Prism (version 9.3.1, GraphPad, USA). Shapiro–Wilk
normality test was performed to confirm the normal distribution of the data using JASP (version 0.16.1, University of Amsterdam, Amsterdam, The Netherlands). The power and sample size were
defined by the Web-based Sample Size/Power Calculator (provided by Dr. Rollin Brant, University of British Columbia, Canada) using the standard deviations calculated from at least three
independent pilot experiments/cohorts of animals. A power of 80% and a significance of 5% were selected. The variances between compared groups were similar. Unpaired _t_-tests were used to
compare the means of two independent groups. One-way ANOVA with Tukey’s multiple comparison were used to determine the differences between three of more independent groups. 2-way ANOVA tests
with Tukey multiple comparisons were used to determine the differences of three or more groups with two independent variables. For tests between groups with repeated measurements mixed
model ANOVA analysis for repeated measurement with Tukey’s multiple comparison was used. A _p_-value ≤ 0.05 was considered statistically significant. DATA AVAILABILITY All data needed to
evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the corresponding author. REFERENCES * International Diabetes
Federation, Brussels, Belgium. IDF diabetes atlas, 10th ed. IDF. 2021. * Pereira SS, Alvarez-Leite JI. Low-grade inflammation, obesity, and diabetes. Curr Obes Rep. 2014;3:422–31. Article
PubMed Google Scholar * Rodriguez-Calvo T, Richardson SJ, Pugliese A. Pancreas pathology during the natural history of type 1 diabetes. Curr Diab Rep. 2018;18:124. Article PubMed Google
Scholar * Patel S, Santani D. Role of NF-kappa B in the pathogenesis of diabetes and its associated complications. Pharm Rep. 2009;61:595–603. Article CAS Google Scholar * Barbe-Tuana
FM, Klein D, Ichii H, Berman DM, Coffey L, Kenyon NS, et al. CD40-CD40 ligand interaction activates proinflammatory pathways in pancreatic islets. Diabetes 2006;55:2437–45. Article CAS
PubMed Google Scholar * Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–26. Article CAS PubMed
Google Scholar * Meyerovich K, Ortis F, Cardozo AK. The non-canonical NF-kappaB pathway and its contribution to beta-cell failure in diabetes. J Mol Endocrinol. 2018;61:F1–F6. Article
CAS PubMed Google Scholar * Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17:545–58. Article CAS PubMed PubMed Central Google Scholar
* Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell 2017;168:37–57. Article CAS PubMed PubMed Central Google Scholar *
Halvorsen B, Santilli F, Scholz H, Sahraoui A, Gulseth HL, Wium C, et al. LIGHT/TNFSF14 is increased in patients with type 2 diabetes mellitus and promotes islet cell dysfunction and
endothelial cell inflammation in vitro. Diabetologia 2016;59:2134–44. Article CAS PubMed PubMed Central Google Scholar * El-Asrar MA, Adly AA, Ismail EA. Soluble CD40L in children and
adolescents with type 1 diabetes: relation to microvascular complications and glycemic control. Pediatr Diabetes. 2012;13:616–24. Article CAS PubMed Google Scholar * Meyerovich K, Fukaya
M, Terra LF, Ortis F, Eizirik DL, Cardozo AK. The non-canonical NF-kappaB pathway is induced by cytokines in pancreatic beta cells and contributes to cell death and proinflammatory
responses in vitro. Diabetologia 2016;59:512–21. Article CAS PubMed Google Scholar * Li X, Wu Y, Song Y, Ding N, Lu M, Jia L, et al. Activation of NF-kappaB-inducing kinase in islet beta
cells causes beta cell failure and diabetes. Mol Ther. 2020;28:2430–41. Article CAS PubMed PubMed Central Google Scholar * Malle EK, Zammit NW, Walters SN, Koay YC, Wu J, Tan BM, et
al. Nuclear factor kappaB-inducing kinase activation as a mechanism of pancreatic beta cell failure in obesity. J Exp Med. 2015;212:1239–54. Article CAS PubMed PubMed Central Google
Scholar * Pflug KM, Sitcheran R. Targeting NF-kappaB-inducing kinase (NIK) in immunity, inflammation, and cancer. Int J Mol Sci. 2020;21. * Sun SC. Non-canonical NF-kappaB signaling
pathway. Cell Res. 2011;21:71–85. Article CAS PubMed Google Scholar * Ray MK, Fagan SP, Moldovan S, DeMayo FJ, Brunicardi FC. A mouse model for beta cell-specific ablation of target
gene(s) using the Cre-loxP system. Biochem Biophys Res Commun. 1998;253:65–9. Article CAS PubMed Google Scholar * Tchoghandjian A, Jennewein C, Eckhardt I, Rajalingam K, Fulda S.
Identification of non-canonical NF-kappaB signaling as a critical mediator of Smac mimetic-stimulated migration and invasion of glioblastoma cells. Cell Death Dis. 2013;4:e564. Article CAS
PubMed PubMed Central Google Scholar * Xiao G, Fong A, Sun SC. Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and
IKKalpha-mediated phosphorylation. J Biol Chem. 2004;279:30099–105. Article CAS PubMed Google Scholar * Dejardin E. The alternative NF-kappaB pathway from biochemistry to biology:
pitfalls and promises for future drug development. Biochem Pharm. 2006;72:1161–79. Article CAS PubMed Google Scholar * McEvoy RC, Andersson J, Sandler S, Hellerstrom C. Multiple low-dose
streptozotocin-induced diabetes in the mouse. Evidence for stimulation of a cytotoxic cellular immune response against an insulin-producing beta cell line. J Clin Investig. 1984;74:715–22.
Article CAS PubMed PubMed Central Google Scholar * Sakata N, Yoshimatsu G, Tsuchiya H, Egawa S, Unno M. Animal models of diabetes mellitus for islet transplantation. Exp Diabetes Res.
2012;2012:256707. Article PubMed PubMed Central CAS Google Scholar * Nackiewicz D, Dan M, Speck M, Chow SZ, Chen YC, Pospisilik JA, et al. Islet macrophages shift to a reparative state
following pancreatic beta-cell death and are a major source of islet insulin-like growth factor-1. iScience. 2020;23:100775. Article CAS PubMed Google Scholar * Kolb H. Mouse models of
insulin dependent diabetes: low-dose streptozocin-induced diabetes and nonobese diabetic (NOD) mice. Diabetes Metab Rev. 1987;3:751–78. Article CAS PubMed Google Scholar * Parel Y,
Chizzolini C. CD4+ CD8+ double positive (DP) T cells in health and disease. Autoimmun Rev. 2004;3:215–20. Article PubMed Google Scholar * Tsonkova VG, Sand FW, Wolf XA, Grunnet LG,
Kirstine Ringgaard A, Ingvorsen C, et al. The EndoC-betaH1 cell line is a valid model of human beta cells and applicable for screenings to identify novel drug target candidates. Mol Metab.
2018;8:144–57. Article CAS PubMed Google Scholar * Gurgul-Convey E, Mehmeti I, Plotz T, Jorns A, Lenzen S. Sensitivity profile of the human EndoC-betaH1 beta cell line to proinflammatory
cytokines. Diabetologia 2016;59:2125–33. Article CAS PubMed Google Scholar * Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The lymphotoxin-beta receptor induces
different patterns of gene expression via two NF-kappaB pathways. Immunity 2002;17:525–35. Article CAS PubMed Google Scholar * Brightbill HD, Suto E, Blaquiere N, Ramamoorthi N,
Sujatha-Bhaskar S, Gogol EB, et al. NF-kappaB inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat Commun. 2018;9:179. Article PubMed PubMed Central CAS Google
Scholar * Maracle CX, Kucharzewska P, Helder B, van der Horst C, Correa de Sampaio P, Noort AR, et al. Targeting non-canonical nuclear factor-kappaB signalling attenuates neovascularization
in a novel 3D model of rheumatoid arthritis synovial angiogenesis. Rheumatology (Oxford). 2017;56:294–302. Article CAS Google Scholar * Donath MY, Dalmas E, Sauter NS, Boni-Schnetzler M.
Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell Metab. 2013;17:860–72. Article CAS PubMed Google Scholar * Evangelou M, Smyth DJ, Fortune MD,
Burren OS, Walker NM, Guo H, et al. A method for gene-based pathway analysis using genomewide association study summary statistics reveals nine new type 1 diabetes associations. Genet
Epidemiol. 2014;38:661–70. Article PubMed PubMed Central Google Scholar * Sever D, Hershko-Moshe A, Srivastava R, Eldor R, Hibsher D, Keren-Shaul H, et al. NF-kappaB activity during
pancreas development regulates adult beta-cell mass by modulating neonatal beta-cell proliferation and apoptosis. Cell Death Discov. 2021;7:2. Article CAS PubMed PubMed Central Google
Scholar * Rossini AA, Williams RM, Appel MC, Like AA. Complete protection from low-dose streptozotocin-induced diabetes in mice. Nature 1978;276:182–4. Article CAS PubMed Google Scholar
* Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science 1976;193:415–7. Article CAS PubMed Google Scholar * Blaquiere N, Castanedo
GM, Burch JD, Berezhkovskiy LM, Brightbill H, Brown S, et al. Scaffold-hopping approach to discover potent, selective, and efficacious inhibitors of NF-kappaB inducing kinase. J Med Chem.
2018;61:6801–13. Article CAS PubMed Google Scholar * Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Investig. 2017;127:2881–91. Article PubMed PubMed Central Google
Scholar * Wang F, Sun F, Luo J, Yue T, Chen L, Zhou H, et al. Loss of ubiquitin-conjugating enzyme E2 (Ubc9) in macrophages exacerbates multiple low-dose streptozotocin-induced diabetes by
attenuating M2 macrophage polarization. Cell Death Dis. 2019;10:892. Article CAS PubMed PubMed Central Google Scholar * Luo Z, Solang C, Mejia-Cordova M, Thorvaldson L, Blix M, Sandler
S, et al. Kinetics of immune cell responses in the multiple low-dose streptozotocin mouse model of type 1 diabetes. FASEB BioAdv. 2019;1:538–49. Article CAS PubMed PubMed Central Google
Scholar * Zhang N, Schroppel B, Lal G, Jakubzick C, Mao X, Chen D, et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune
response. Immunity 2009;30:458–69. Article CAS PubMed PubMed Central Google Scholar * Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, et al. Visualizing regulatory T cell control
of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92. Article CAS PubMed Google Scholar * Clenet ML, Gagnon F, Moratalla AC, Viel EC, Arbour N. Peripheral human
CD4(+)CD8(+) T lymphocytes exhibit a memory phenotype and enhanced responses to IL-2, IL-7 and IL-15. Sci Rep. 2017;7:11612. Article PubMed PubMed Central CAS Google Scholar *
Kucharzewska P, Maracle CX, Jeucken KCM, van Hamburg JP, Israelsson E, Furber M, et al. NIK–IKK complex interaction controls NF-kappaB-dependent inflammatory activation of endothelium in
response to LTbetaR ligation. J Cell Sci. 2019;132. * Al-Sadi R, Guo S, Ye D, Rawat M, Ma TY. TNF-alpha modulation of intestinal tight junction permeability is mediated by NIK/IKK-alpha axis
activation of the canonical NF-kappaB pathway. Am J Pathol. 2016;186:1151–65. Article CAS PubMed PubMed Central Google Scholar * Zarnegar B, Yamazaki S, He JQ, Cheng G. Control of
canonical NF-kappaB activation through the NIK–IKK complex pathway. Proc Natl Acad Sci USA. 2008;105:3503–8. Article CAS PubMed PubMed Central Google Scholar * Sheng L, Zhou Y, Chen Z,
Ren D, Cho KW, Jiang L, et al. NF-kappaB-inducing kinase (NIK) promotes hyperglycemia and glucose intolerance in obesity by augmenting glucagon action. Nat Med. 2012;18:943–9. Article CAS
PubMed PubMed Central Google Scholar * Yoshimatsu G, Kunnathodi F, Saravanan PB, Shahbazov R, Chang C, Darden CM, et al. Pancreatic beta-cell-derived IP-10/CXCL10 isletokine mediates
early loss of graft function in islet cell transplantation. Diabetes 2017;66:2857–67. Article CAS PubMed PubMed Central Google Scholar * Cardozo AK, Proost P, Gysemans C, Chen MC,
Mathieu C. Eizirik DLIL-1beta and IFN-gamma induce the expression of diverse chemokines and IL-15 in human and rat pancreatic islet cells, and in islets from pre-diabetic NOD mice.
Diabetologia. 2003;46:255–66. Article CAS PubMed Google Scholar * Frigerio S, Junt T, Lu B, Gerard C, Zumsteg U, Hollander GA, et al. Beta cells are responsible for CXCR3-mediated T-cell
infiltration in insulitis. Nat Med. 2002;8:1414–20. Article CAS PubMed Google Scholar * Grieco FA, Moore F, Vigneron F, Santin I, Villate O, Marselli L, et al. IL-17A increases the
expression of proinflammatory chemokines in human pancreatic islets. Diabetologia. 2014;57:502–11. Article CAS PubMed Google Scholar * Cardozo AK, Ortis F, Storling J, Feng YM,
Rasschaert J, Tonnesen M, et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic
reticulum stress in pancreatic beta-cells. Diabetes 2005;54:452–61. Article CAS PubMed Google Scholar * Ravassard P, Hazhouz Y, Pechberty S, Bricout-Neveu E, Armanet M, Czernichow P, et
al. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J Clin Investig. 2011;121:3589–97. Article CAS PubMed PubMed Central Google
Scholar * Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic
islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002;51:1437–42. Article CAS PubMed Google Scholar *
Allagnat F, Fukaya M, Nogueira TC, Delaroche D, Welsh N, Marselli L, et al. C/EBP homologous protein contributes to cytokine-induced pro-inflammatory responses and apoptosis in beta-cells.
Cell Death Differ. 2012;19:1836–46. Article CAS PubMed PubMed Central Google Scholar * Moore F, Cunha DA, Mulder H, Eizirik DL. Use of RNA interference to investigate cytokine signal
transduction in pancreatic beta cells. Methods Mol Biol. 2012;820:179–94. Article CAS PubMed Google Scholar * Schaffer AE, Yang AJ, Thorel F, Herrera PL, Sander M. Transgenic
overexpression of the transcription factor Nkx6.1 in beta-cells of mice does not increase beta-cell proliferation, beta-cell mass, or improve glucose clearance. Mol Endocrinol.
2011;25:1904–14. Article CAS PubMed PubMed Central Google Scholar * Hennige AM, Burks DJ, Ozcan U, Kulkarni RN, Ye J, Park S, et al. Upregulation of insulin receptor substrate-2 in
pancreatic beta cells prevents diabetes. J Clin Investig. 2003;112:1521–32. Article CAS PubMed PubMed Central Google Scholar * Meyerovich K, Violato NM, Fukaya M, Dirix V, Pachera N,
Marselli L, et al. MCL-1 is a key antiapoptotic protein in human and rodent pancreatic beta-cells. Diabetes 2017;66:2446–58. Article CAS PubMed Google Scholar * Marroqui L, Masini M,
Merino B, Grieco FA, Millard I, Dubois C, et al. Pancreatic alpha cells are resistant to metabolic stress-induced apoptosis in type 2 diabetes. EBioMedicine 2015;2:378–85. Article PubMed
PubMed Central Google Scholar * Fukaya M, Brorsson CA, Meyerovich K, Catrysse L, Delaroche D, Vanzela EC, et al. A20 inhibits beta-cell apoptosis by multiple mechanisms and predicts
residual beta-cell function in type 1 diabetes. Mol Endocrinol. 2016;30:48–61. Article CAS PubMed Google Scholar * Wagner AM, Cloos P, Bergholdt R, Eising S, Brorsson C, Stalhut M, et
al. Posttranslational protein modifications in type 1 diabetes—genetic studies with PCMT1, the repair enzyme protein isoaspartate methyltransferase (PIMT) encoding gene. Rev Diabet Stud.
2008;5:225–31. Article PubMed Google Scholar * Catrysse L, Fukaya M, Sze M, Meyerovich K, Beyaert R, Cardozo AK, et al. A20 deficiency sensitizes pancreatic beta cells to cytokine-induced
apoptosis in vitro but does not influence type 1 diabetes development in vivo. Cell Death Dis. 2015;6:e1918. Article CAS PubMed PubMed Central Google Scholar * Takiishi T, Cook DP,
Korf H, Sebastiani G, Mancarella F, Cunha JP, et al. Reversal of diabetes in NOD mice by clinical-grade proinsulin and IL-10-secreting _Lactococcus lactis_ in combination with low-dose
anti-CD3 depends on the induction of Foxp3-positive T cells. Diabetes 2017;66:448–59. Article CAS PubMed Google Scholar * Rasschaert J, Ladriere L, Urbain M, Dogusan Z, Katabua B, Sato
S, et al. Toll-like Receptor 3 and STAT-1 contribute to double-stranded RNA+ interferon-gamma-induced apoptosis in primary pancreatic beta-cells. J Biol Chem. 2005;280:33984–91. Article CAS
PubMed Google Scholar * Allagnat F, Cunha D, Moore F, Vanderwinden JM, Eizirik DL, Cardozo AK. Mcl-1 downregulation by pro-inflammatory cytokines and palmitate is an early event
contributing to beta-cell apoptosis. Cell Death Differ. 2011;18:328–37. Article CAS PubMed Google Scholar * Mortier J, Frederick R, Ganeff C, Remouchamps C, Talaga P, Pochet L, et al.
Pyrazolo[4,3-c]isoquinolines as potential inhibitors of NF-kappaB activation. Biochem Pharm. 2010;79:1462–72. Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We
thank A. Musuaya, C. Dubois and M. Popa for technical assistance. Work in the AKC and ED groups were supported by the Excellence of Science grant (FNRS, Belgium, convention 30826052). FD is
supported by the Italian Ministry of University and Research (2268-2019-DF-CONRICMIUR PRIN2017_001) and by the Italian Ministry of Health (PROMETEO). GS was supported by the Italian Ministry
of University and Research (201793XZ5A_006) and by the Italian Ministry of Health “Ricerca Finalizzata 2018” (GR-2018-12365577). Work in SPS’s lab was supported by MISU (34772792) and
MISU-PROL (40005588) funding from the FNRS and from Fondation Jaumotte-Demoulin. ENG is supported by a Fonds National de la Recherche Scientifique (FNRS)-MIS grant (33650793), an FNRS-CDR
grant (35275350), a European Research Council (ERC) Consolidator grant METAPTPs (GA817940), and a JDRF Career Development Award (CDA-2019-758-A-N). ENG is a Research Associate of the FNRS,
Belgium. AUTHOR INFORMATION Author notes * These authors contributed equally: Peng Xiao, Tatiana Takiishi. AUTHORS AND AFFILIATIONS * Inflammation and Cell Death Signalling group,
Laboratoire de Gastroentérologie Expérimental et Endotools, Université libre de Bruxelles, Brussels, Belgium Peng Xiao, Tatiana Takiishi, Natalia Moretti Violato & Alessandra Kupper
Cardozo * Department of Medical Sciences, Surgery and Neurosciences, University of Siena, Siena, Italy Giada Licata, Francesco Dotta & Guido Sebastiani * Fondazione Umberto Di Mario, c/o
Toscana Life Sciences, Siena, Italy Giada Licata, Francesco Dotta & Guido Sebastiani * Tuscany Centre for Precision Medicine (CReMeP), Siena, Italy Francesco Dotta * Department of
Clinical and Experimental Medicine, Islet Laboratory, University of Pisa, Pisa, Italy Lorella Marselli * Institute for Interdisciplinary Research in Human and Molecular Biology, Medical
Faculty, Université libre de Bruxelles, Brussels, Belgium Sumeet Pal Singh * Center for Inflammation Research, VIB, B-9052, Ghent, Belgium Mozes Sze & Geert Van Loo * Department of
Biomedical Molecular Biology, Ghent University, B-9052, Ghent, Belgium Mozes Sze & Geert Van Loo * Laboratory of Molecular Immunology and Signal Transduction, GIGA-Insitute, ULiege,
Liège, Belgium Emmanuel Dejardin * Signal Transduction and Metabolism Laboratory, Laboratoire de Gastroentérologie Expérimental et Endotools, Université libre de Bruxelles, Brussels, Belgium
Esteban Nicolas Gurzov Authors * Peng Xiao View author publications You can also search for this author inPubMed Google Scholar * Tatiana Takiishi View author publications You can also
search for this author inPubMed Google Scholar * Natalia Moretti Violato View author publications You can also search for this author inPubMed Google Scholar * Giada Licata View author
publications You can also search for this author inPubMed Google Scholar * Francesco Dotta View author publications You can also search for this author inPubMed Google Scholar * Guido
Sebastiani View author publications You can also search for this author inPubMed Google Scholar * Lorella Marselli View author publications You can also search for this author inPubMed
Google Scholar * Sumeet Pal Singh View author publications You can also search for this author inPubMed Google Scholar * Mozes Sze View author publications You can also search for this
author inPubMed Google Scholar * Geert Van Loo View author publications You can also search for this author inPubMed Google Scholar * Emmanuel Dejardin View author publications You can also
search for this author inPubMed Google Scholar * Esteban Nicolas Gurzov View author publications You can also search for this author inPubMed Google Scholar * Alessandra Kupper Cardozo View
author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS PX, TT, FD, GS, ED, ENG and AKC contributed to the study concept and design, analysis and
interpretation of the data. PX, TT and NVM contributed to the acquisition of the data. LM, SPS, MS and GVL contributed reagents/materials/analytical tools. PX, TT and AKC wrote/edited the
manuscript. AKC is responsible for its content. All authors revised the article and approved the final version. CORRESPONDING AUTHOR Correspondence to Alessandra Kupper Cardozo. ETHICS
DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICS STATEMENT Mice were housed and handled according to the Belgian Regulations for Animal Care and with
permission from the local Ethic Committee (627N). The human islets (from cadaveric organ donors) are obtained through a scientific collaboration with Prof. Lorela Marselli of the University
of Pisa. An informed consent was obtained and the ethical approval for the work has been granted by the Ethics Committee of the University of Pisa, Italy. ADDITIONAL INFORMATION PUBLISHER’S
NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by Alessandra K. Cardozo SUPPLEMENTARY INFORMATION
REPRODUCIBILITY CHECKLIST SUPPLEMENTAL DATA ORIGINAL DATA FILE RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License,
which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link
to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless
indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or
exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints
and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Xiao, P., Takiishi, T., Violato, N.M. _et al._ NF-κB-inducing kinase (NIK) is activated in pancreatic β-cells but does not contribute to
the development of diabetes. _Cell Death Dis_ 13, 476 (2022). https://doi.org/10.1038/s41419-022-04931-5 Download citation * Received: 22 February 2022 * Revised: 04 May 2022 * Accepted: 09
May 2022 * Published: 19 May 2022 * DOI: https://doi.org/10.1038/s41419-022-04931-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative