The downregulation of type i ifn signaling in g-mdscs under tumor conditions promotes their development towards an immunosuppressive phenotype
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Tumors modify myeloid cell differentiation and induce an immunosuppressive microenvironment. Granulocytic myeloid-derived suppressor cells (G-MDSCs), the main subgroup of
myeloid-derived suppressor cells (MDSCs), are immature myeloid cells (IMCs) with immunosuppressive activity and exist in tumor-bearing hosts. The reason why these cells diverge from a normal
differentiation pathway and are shaped into immunosuppressive cells remains unclear. Here, we reported that the increase of granulocyte colony-stimulating factor (G-CSF) in mouse serum with
tumor progression encouraged G-MDSCs to obtain immunosuppressive traits in peripheral blood through the PI3K-Akt/mTOR pathway. Importantly, we found that downregulation of type I interferon
(IFN-I) signaling in G-MDSCs was a prerequisite for their immunosuppressive effects. Suppressor of cytokine signaling (SOCS1), the action of which is dependent on IFN-I signaling, inhibited
the activation of the PI3K-Akt/mTOR pathway by directly interacting with Akt, indicating that the differentiation of immunosuppressive G-MDSCs involves a transition from immune activation
to immune tolerance. Our study suggests that increasing IFN-I signaling in G-MDSCs may be a strategy for reprograming immunosuppressive myelopoiesis and slowing tumor progression. SIMILAR
CONTENT BEING VIEWED BY OTHERS IMMUNE SUPPRESSIVE ACTIVITY OF MYELOID-DERIVED SUPPRESSOR CELLS IN CANCER REQUIRES INACTIVATION OF THE TYPE I INTERFERON PATHWAY Article Open access 19 March
2021 EXPORTIN 1 GOVERNS THE IMMUNOSUPPRESSIVE FUNCTIONS OF MYELOID-DERIVED SUPPRESSOR CELLS IN TUMORS THROUGH ERK1/2 NUCLEAR EXPORT Article 20 June 2024 MYELOID-DERIVED SUPPRESSOR CELLS AS
IMMUNOSUPPRESSIVE REGULATORS AND THERAPEUTIC TARGETS IN CANCER Article Open access 07 October 2021 INTRODUCTION Avoiding immune destruction is an important characteristic of cancer cells
[1]. Tumor conditions can induce the generation of multiple immunosuppressive cells (regulatory T cells (Tregs), tumor-associated macrophages, myeloid-derived suppressor cells (MDSCs), etc.)
[2]. MDSCs have a phenotype like that of immature myeloid cells (IMCs). Under physiological conditions, IMCs differentiate into dendritic cells, macrophages, and granulocytes. However,
MDSCs can abnormally accumulate under chronic inflammatory conditions, cancer, or autoimmunity [3]. MDSCs have been shown to inhibit antitumor immunity [4]. Upregulation of immunosuppressive
molecules (such as Arg1, iNOS, and TGF-β) in MDSCs leads to suppression of T-cell proliferation and function [5]. MDSCs can also secrete some cytokines to promote tumor cells survival,
proliferation and metastasis [6, 7]. In cancer, MDSC expansion and differentiation are associated with cytokines secreted by tumor cells and tumor stroma cells [8,9,10]. Proinflammatory
mediators, such as IL-1β, IL-6, and PGE2, affect the normal differentiation of IMCs and regulate MDSC accumulation [11]. MDSCs consist of two populations: monocytic MDSCs (mo-MDSCs as
CD11b+Ly6ChiLy6G–) and granulocytic MDSCs (G-MDSCs as CD11b+Ly6CloLy6G+) in mice [6]. The proportions of the two MDSC subtypes are different, and ~70–80% are G-MDSCs [12,13,14,15]. One more
important point is that G-MDSCs are generally much more prevalent than mo-MDSCs in cancer [16]. Type I interferon (IFN-I) is the central antiviral cytokine that transmits signals through the
widely expressed IFNAR receptor (consisting of two subunits, IFNAR1 and IFNAR2) [17]. IFN-I can induce the expression of genes called interferon-stimulated genes (ISGs). ISGs exert
antiviral effects via their immunomodulatory properties or by directly interfering with viral replication [18]. Moreover, IFN-I promotes immune maturation and differentiation from an innate
to an adaptive immune response [19]. Recent work showed that immunosuppressive activity of MDSCs in cancer required inactivation of IFN-I pathway [20]. Although it has been reported that
downregulation of IFN-I signaling is closely related to tumor development [21,22,23,24], little is known about the mechanisms between IFN-I signaling in MDSCs and their immunosuppressive
activity. Although the function of MDSCs has been clearly studied, the development of MDSCs under tumor conditions remains to be elucidated. In fact, where and how IMCs differentiate into
MDSCs are not known in detail [25, 26]. Here, we sought to clarify the process by which G-MDSCs, the main subgroup of MDSCs, obtain immunosuppressive functions and explore the mechanism of
their functional transformation with tumor progression. We reported that the immunosuppressive functions of G-MDSCs were first obtained in the peripheral blood of tumor-bearing mice.
Upregulated G-CSF in the serum was involved in the acquisition of immunosuppressive functions by G-MDSCs via PI3K-Akt/mTOR pathway. Most importantly, the downregulation of IFN-I signaling in
G-MDSCs led to the activation of the PI3K-Akt/mTOR pathway, and SOCS1 which is dependented on IFN-I signaling regulated the PI3K-Akt/mTOR pathway via direct binding of Akt. These results
indicate that the downregulation of IFN-I signaling in G-MDSCs is necessary to obtain immunosuppressive function, suggesting that upregulation of IFN-I signaling may attenuate tumor
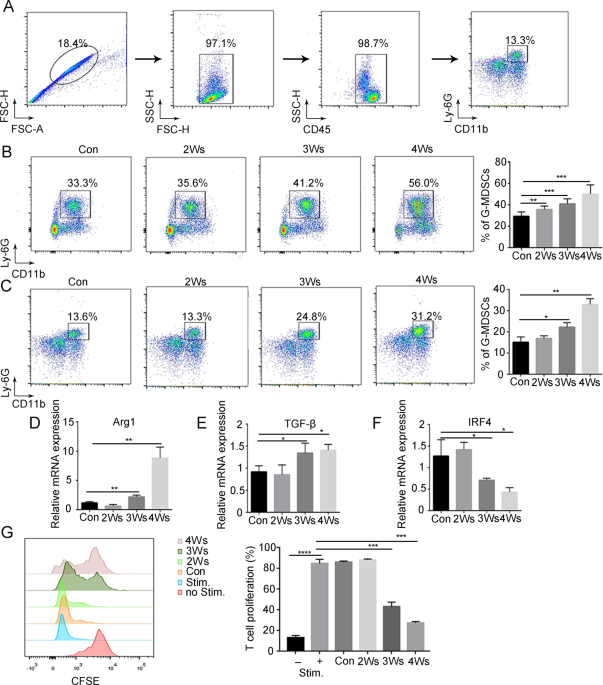
progression. RESULTS G-MDSCS ARE ACTIVATED IN PERIPHERAL BLOOD DURING TUMOR PROGRESSION To address that abnormal amplification and activation of G-MDSCs under tumor conditions, B16F10
melanoma cells were injected subcutaneously into mice. The number of G-MDSCs was examined by flow cytometry (Fig. 1A). With tumor progression, G-MDSCs accumulated in bone marrow and
peripheral blood (Fig. 1B, C and Supplementary Fig. 1A). To determine whether G-MDSCs have an immunosuppressive function in the early stage of development, the immunosuppressive activity of
bone marrow G-MDSCs was examined. The results showed that the expression of the immunosuppressive genes Arg1 and TGFβ in bone marrow G-MDSCs did not change with tumor progression
(Supplementary Fig. 1B). Moreover, bone marrow G-MDSCs showed no inhibition of CD8+ T-cell proliferation (Supplementary Fig. 1C). These results suggest that bone marrow G-MDSCs have no
immunosuppressive function, which means that bona fide G-MDSCs require further activation. Next, the immunosuppressive activity of peripheral blood G-MDSCs was examined. The expression of
Arg1 and TGF-β in peripheral blood G-MDSCs began to significantly increase after 3 weeks of tumor bearing (Fig. 1D, E). IRF4 negatively regulates the immunosuppressive function of MDSCs [27]
and IRF4 in peripheral blood G-MDSCs was significantly decreased after 3 weeks of tumor bearing (Fig. 1F). Then we found that peripheral blood G-MDSCs of mice bearing tumors for 3 weeks
significantly inhibited T-cell proliferation (Fig. 1G), indicating that G-MDSCs acquire immunosuppressive functions in the peripheral blood. CXCR2 and CD101 can be used as markers for the
maturity of polymorphonuclear cells [28]. Our results showed that the expression of CXCR2 and CD101 in peripheral blood G-MDSCs of tumor-bearing mice was reduced (Supplementary Fig. 1D, E).
This result indicates that peripheral blood G-MDSCs are in an immature stage. ACCUMULATION OF G-MDSCS WITH IMMUNOSUPPRESSIVE FUNCTIONS PROMOTES TUMOR GROWTH AND METASTASIS Polymorphonuclear
cells are considered to be short-lived cells [29]. We found that the apoptosis of peripheral blood G-MDSCs was decreased under tumor conditions (Fig. 2A), thus contributing to G-MDSC
activity. The tumor tissue is the final location in which G-MDSCs exert immunosuppressive functions [30]. We confirmed that G-MDSCs could be recruited into tumor tissue (Fig. 2B). To detect
the effect of G-MDSCs on tumor growth and metastasis, peripheral blood G-MDSCs of normal mice or mice bearing tumors for 3 weeks were injected into mice by tail vein (Fig. 2C). In the group
of mice injected with peripheral blood G-MDSCs of mice bearing tumors for 3 weeks, the number of G-MDSCs in peripheral blood and tumor tissue was significantly higher than that in other
groups (Fig. 2D, E and Supplementary Fig. 2A), and a significant increase in tumor growth was observed (Fig. 2F). G-MDSCs with immunosuppressive functions also promoted tumor cells
metastasis to the lung (Fig. 2G, H). After G-MDSCs were depleted by anti-GR1 antibody (Fig. 2I, J, L and Supplementary Fig. 2B), tumor growth and metastasis were inhibited (Fig. 2K, M).
Overall, these results indicate that immunosuppressive G-MDSCs promote tumor growth and metastasis. ACTIVATION OF THE PI3K-AKT/MTOR PATHWAY IS RELATED TO THE EXPRESSION OF IMMUNOSUPPRESSIVE
GENE To explore the nature of IMC differentiation into immunosuppressive G-MDSCs, transcriptome sequencing was performed. The purity of separated G-MDSCs was detected (Supplementary Fig.
3A). Differential expression analysis of the sequencing data revealed the gene expression patterns of peripheral blood G-MDSCs from tumor-bearing mice at different stages. With tumor
progression, the number of genes that changed in G-MDSCs gradually increased (Fig. 3A). Kyoto Encyclopedia of Genes and Genomes pathway analysis performed on RNA-seq data revealed an
enrichment of the PI3K-Akt pathway in immunosppressive G-MDSCs (Fig. 3B). PI3K-Akt signaling regulates metabolism, growth and survival through mammalian target of rapamycin (mTOR), and it
has also been recognized that the PI3K-Akt/mTOR pathway has broad roles in immune cells [31, 32]. Indeed, we observed that the phosphorylation of Akt and mTOR was upregulated in
immunosppressive G-MDSCs (Fig. 3C). mTOR can further activate Stat3, which plays an important role in immunosuppression [33,34,35], suggesting that the PI3K-Akt/mTOR pathway is likely a
driver of G-MDSC function. We next examined the effect of inhibitors (an mTOR inhibitor, rapamycin, and a Stat3 inhibitor, stattic) on G-MDSCs in vivo, and the results showed that the
percentage of G-MDSCs in peripheral blood and the expression of Arg1 both decreased after injecting rapamycin (Fig. 3D–F and Supplementary Fig. 3B). Taken together, these results indicate
that the PI3K-Akt/mTOR pathway promotes G-MDSCs to obtain immunosuppressive function. IFN-I SIGNALING IS INHIBITED AS G-MDSCS DIFFERENTIATE TOWARD AN IMMUNOSUPPRESSIVE PHENOTYPE IFN-I has a
pivotal role in promoting antitumor responses, and IFN-I signaling is essential for antiviral and immunoregulatory actions via induction of ISGs [36, 37]. The transcriptome sequencing
results showed that ISGs were significantly downregulated during G-MDSCs differentiation. Gene Ontology enrichment analysis demonstrated that the differentially expressed genes (DEGs) were
mainly related to the IFN-I response (Fig. 4A, B). The results were validated by Q-PCR (Fig. 4C). Downregulation of ISGs could be mapped at multiple levels in the IFN-JAK-STAT1 signal
cascade (Fig. 4D). The content of IFNα was detected by ELISA. Strikingly, we observed an obvious upregulation of IFNα in serum from mice bearing tumors for 3 weeks (Fig. 4E). The loss of
IFNAR could also result in the downregulation of ISGs. Indeed, IFNAR was significantly downregulated in immunosuppressive G-MDSCs (Fig. 4F, G). And the protein level of IFNAR and the Stat1
phosphorylation in G-MDSCs also decreased (Fig. 4H). The relationship between the downregulation of IFN-I signaling and the immunosuppressive function of G-MDSCs was next investigated. The
expression of Arg1 in G-MDSCs from different tissues was detected, and the results showed that Arg1 expression was gradually upregulated in bone marrow, peripheral blood and tumor tissue
(Supplementary Fig. 4A). We then examined IFN-I signaling expression in G-MDSCs from different tissues, and the results showed that G-MDSCs from tumor tissue had the strongest
immunosuppressive activity and the lowest IFNAR (Supplementary Fig. 4B, C) and ISGs expression (Supplementary Fig. 4D–G). In vitro stimulation with IFNα showed that the G-MDSCs from tumor
tissue barely responded to IFNα (Supplementary Fig. 4H–M). These results indicate that the IFN-I signaling of immunosuppressive G-MDSCs is inhibited. G-CSF IS REQUIRED FOR THE ACTIVATION OF
PI3K-AKT/MTOR PATHWAY AND G-MDSC IMMUNOSUPPRESSIVE FUNCTION To determine the relationship between G-MDSC immunosuppressive function and tumor progression, we used conditioned medium from
nontumor cells (mouse embryonic fibroblasts (MEFs)) and B16F10 and serum from tumor-bearing mice to induce G-MDSC differentiation. The results showed that the Arg1 expression in bone marrow
G-MDSCs induced by tumor conditioned medium and serum from tumor-bearing mice was significantly upregulated (Fig. 5A). To identify the factors associated with this process, we analyzed the
serum of mice bearing tumors for 0, 2 or 3 weeks by using a proteome profiling array. CXCL10, CXCL13, and G-CSF were present at higher concentrations in the serum of tumor-bearing mice (Fig.
5B). The source of the changed factors was detected, and the results showed that B16F10 expressed CXCL10 and CXCL13, but G-CSF was secreted by other stromal cells (Supplementary Fig. 5A).
G-MDSCs expressed all the receptors of CXCL10, CXCL13, and G-CSF (CXCR3, CXCR5, and G-CSFR, respectively) (Supplementary Fig. 5B). Then, the effect of altered cytokines on the
immunosuppressive function of G-MDSCs was determined in vitro. The results showed that only G-CSF increased the expression of Arg1 (Fig. 5C), and CXCL10 and CXCL13 could not directly affect
the expression of Arg1 (Supplementary Fig. 5C, D). We confirmed the upregulation of G-CSF in serum of tumor mice by ELISA (Supplementary Fig. 5E). Then we generated two independent short
hairpin RNAs (shRNAs) to stably decrease G-CSF levels (Fig. 5D and Supplementary Fig. 5F). After shRNA transfection in vivo, we observed a decrease in the number of G-MDSCs and a decrease in
the expression of Arg1 (Fig. 5E, F and Supplementary Fig. 5G). Next, the effect of G-CSF on the PI3K-Akt/mTOR pathway was examined. After treatment of bone marrow G-MDSCs with G-CSF in
vitro, PI3K-Akt/mTOR pathway-related proteins were activated (Fig. 5G). The effect of rapamycin on G-CSF-stimulated G-MDSCs was next investigated, and the results showed that after treatment
with rapamycin, Stat3 phosphorylation decreased and Arg1 expression was decreased (Fig. 5H, I). Consistent with previous reports [34, 38, 39], stattic treatment reduced the expression of
Arg1 (Fig. 5J, K). These results suggest that G-CSF promotes G-MDSCs immunosuppressive functions through PI3K-Akt/mTOR pathway. DOWNREGULATION OF IFN-I SIGNALING IS A PREREQUISITE FOR
G-MDSCS TO OBTAIN IMMUNOSUPPRESSIVE FUNCTION We then explored whether G-CSF affects IFN-I signaling in G-MDSCs. The expression of IFNAR and ISGs in bone marrow G-MDSCs was detected after
stimulation with G-CSF in vitro. The results showed that G-CSF could not downregulate the expression of IFNAR (Supplementary Fig. 6A, B) and ISGs (Supplementary Fig. 6C–F). When stimulated
with the serum of tumor-bearing mice, G-MDSCs showed significantly downregulated expression of IFNAR (Fig. 6A, B) and ISGs (Fig. 6C–F). Because the ISGs expression in G-MDSCs began to
decrease after 2 weeks of tumor bearing, we speculated that downregulation of IFN-I signaling may be required for G-MDSCs to obtain immunosuppressive functions. We first stimulated bone
marrow G-MDSCs with IFNα and then stimulated them with G-CSF. The results showed that when G-MDSCs were first stimulated by IFNα, G-CSF did not activate the PI3K-Akt/mTOR pathway (Fig. 6G).
Similarly, the Arg1 expression was inhibited (Fig. 6H). 32D clone 3 is a mouse progenitor cell line that can be induced to differentiate into neutrophils after the addition of stem cell
factor and IL-3 and the subsequent addition of G-CSF in vitro [40, 41]. We evaluated the effects of IFN-I signaling on tumor-induced G-MDSC differentiation, and the scheme of the
experimental design is shown in Fig. 6I. With the differentiation of 32D cells into neutrophils, IFNAR was upregulated (Fig. 6J, K), and the upregulation of IFN-I signaling (Supplementary
Fig. 7A–D) was followed by the downregulation of the PI3K-Akt/mTOR pathway and Arg1 expression (Fig. 6L, M). Interference with IFNAR (Supplementary Fig. 7E, F) attenuated IFN-I signaling
(Supplementary Fig. 7G–J) and thus promoted the upregulation of the PI3K-Akt/mTOR pathway and Arg1 expression (Fig. 6N, O). The above results indicate that the downregulation of IFN-I
signaling in G-MDSCs under tumor conditions promotes their development toward an immunosuppressive phenotype. We also detected the expression of IFN-I signaling in mo-MDSCs, and the results
showed that the IFN-I signaling in mo-MDSCs was also downregulated (Supplementary Fig. 8), suggesting that the downregulation of IFN-I signaling under tumor conditions is necessary for the
formation of an immunosuppressive microenvironment. SOCS1, WHICH IS DEPENDENT ON IFN-I SIGNALING, REGULATES THE ACTIVATION OF THE PI3K-AKT/MTOR PATHWAY Based on the above findings, we
explored the mechanism by which IFN-I signaling regulates PI3K-Akt pathway in G-MDSCs. Suppressor of cytokine signaling (SOCS) members are thought to act as classic negative feedback
inhibitors, which are induced by cytokines, and subsequently inhibit the function of cytokines [42, 43]. SOCS1 can not only regulate IFN-I signaling via negative feedback [17] but also
inhibit the PI3K-Akt pathway [44, 45]. Our results showed that with tumor progression, the expression of SOCS1 in peripheral blood G-MDSCs was gradually downregulated (Fig. 7A and
Supplementary Fig. 9A). We used IFNα to stimulate bone marrow G-MDSCs and 32D cells in vitro to detect the expression of SOCS1, and the results showed that IFNα could significantly induce
the expression of SOCS1 (Fig. 7B, C and Supplementary Fig. 9B, C). Consistently, after interference with IFNAR, the expression of SOCS1 was significantly downregulated (Fig. 7D and
Supplementary Fig. 9D). We speculated that the expression of SOCS1 induced by IFN-I is related to the PI3K-Akt pathway in G-MDSCs. To assess this possibility, we designed siRNA to interfere
with SOCS1 in 32D cells and detected the interference efficiency (Fig. 7E and Supplementary Fig. 9E). Then, the effects of SOCS1 on the PI3K-Akt/mTOR pathway activation and Arg1 expression
were examined. The results showed that the phosphorylation of Akt and mTOR and the expression of Arg1 in 32D cells were upregulated after interference with SOCS1 (Fig. 7F, G). To explore how
SOCS1 inhibits the activation of PI3K-Akt pathway, a co-immunoprecipitation (co-IP) assay was performed. The results showed that SOCS1 and Akt existed in the same complex (Fig. 7H, I). To
confirm whether SOCS1 directly binds to Akt, we prepared His-SOCS1 and GST-Akt constructs from bacteria, and in vitro GST pull-down assays showed that SOCS1 physically associates with Akt
through direct interaction (Fig. 7J). Next, we assessed which domain of SOCS1 interacts with Akt. SOCS1 consists of an N-terminal region, a kinase inhibitory region (KIR), a central SH2
region and a carboxy-terminal SOCS box [46]. We generated His-tagged deletion mutants containing the N-terminal region, N-terminal region-KIR, and N-terminal region-KIR-SH2 domains (Fig.
7K), and pull-down assays were performed. The results showed that GST-Akt could pull-down His-tagged mutants only in the presence of the SOCS1 SH2 domain (Fig. 7L), indicating that the SH2
domain of SOCS1 is required for inhibiting PI3K-Akt/mTOR signaling. DISCUSSION The main function of MDSCs is immunosuppression, but this function is not innate. It has been reported that
MDSCs at the early stage of tumors do not exhibit immunosuppression [47, 48]. It is not clear where and how MDSCs obtain immunosuppressive functions. Here, we reported that G-MDSCs, the main
subgroup of MDSCs, initiated immunosuppressive functions in the peripheral blood under tumor conditions. Our research deeply analyzed the specific processes by which G-MDSCs are activated
and provides new evidence that MDSCs with immunosuppressive functions need to undergo two steps, amplification and activation. Tumor and/or stromal cells secrete inflammatory mediators to
influence myeloid cells differentiation [9, 10, 49]. G-CSF has multiple effects on polymorphonuclear cells, including regulation of proliferation and mobilization [50]. In many tumors, high
levels of G-CSF are produced [50,51,52]. Our results indicate that the upregulation of G-CSF participates in the initiation of G-MDSCs immunosuppressive function. It has been reported that
the absence of PI3Kγ promoted the transformation of macrophages into antitumor M1 macrophages [53], and PI3Kγ signaling promoting immune suppression in cancer [54]. The role of the
PI3K-Akt/mTOR axis in G-MDSC differentiation remains largely unstudied. Here, we showed that the PI3K-Akt/mTOR signaling of peripheral blood G-MDSCs is activated and that G-CSF promotes the
acquisition of immunosuppressive functions by G-MDSCs by activating the PI3K-Akt/mTOR pathway, suggesting that inhibition of PI3K-Akt/mTOR can inhibit G-MDSC immunosuppressive function and
improve immunosuppressive tumor microenvironment. The markers of neutrophils and G-MDSCs are identical. Neutrophils are considered as important effectors of the innate immune system [55]. In
cancer, neutrophils undergo a phenotypic change regarded as “alternating activation” and are called tumor-associated neutrophils (TANs) [56]. Both TANs and G-MDSCs can exert
immunosuppressive functions and are very similar to each other [57]. The only difference between the two may be maturity, however, some studies have pointed out that the suppressive
functions of immature and mature neutrophils is a pathological response to tumorigenesis rather than a completely separate granulocytic population [28]. All told, it is necessary to study
the immunosuppressive function of these cells in depth, and whether these cells are called G-MDSC or TAN is less important. IFN-I is related to the maturity of neutrophils and the
development of immune function [58]. The role of IFN-I in tumor immunosuppression remains controversial. Chronic IFNα promotes MDSC accumulation [59], and IFNα can also promote the
expression of PD-L1 and PD-1, which contributes to immunosuppression [60]. Conversely, stimulator of interferon genes (STING) inhibits MDSC differentiation by activating IFN-I signaling in
Epstein-Barr virus-associated nasopharyngeal carcinoma [61]. Based on our findings, although the IFNα level in mouse serum increased with tumor progression, the IFN-I signaling in G-MDSCs is
downregulated and its downregulation during G-MDSC differentiation was related to the acquisition of immunosuppressive function. We found that SOCS1, a classical negative feedback
regulator, which depends on IFN-I signaling, can directly bind with Akt through SH2 domain to inhibit the immunosuppressive PI3K-Akt/mTOR signaling pathway in G-MDSCs. The downregulation of
IFN-I signaling under tumor conditions leads to the downregulation of SOCS1, which causes the activation of the PI3K-Akt/mTOR pathway. The IFNAR expression is essential for IFN-I signal
transduction. In our study, the IFNAR expression in immunosuppressive G-MDSCs decreased, as a result, the response of G-MDSCs to IFN-I was reduced. Our results indicate that the
downregulation of IFNAR promotes the activation of PI3K-Akt/mTOR, which is related to the immunosuppressive function of G-MDSCs and we further clarifies the specific mechanism by which tumor
conditions induce immune activation to tilt toward immunosuppression. Previous studies have revealed that VEGF or IFN-α/β promotes the ubiquitination and degradation of IFNAR1 and ensuing
restriction of cellular responses to IFN-I [62, 63]. Activation of p38 kinase is also required for IFNAR1 ubiquitination and degradation [64]. In our study, although G-CSF promotes the
initiation of G-MDSCs immunosuppressive function through PI3K-Akt/mTOR pathway, only G-CSF is not enough, G-CSF can not downregulate IFN-I signaling in G-MDSCs. It remains to be elucidated
what factors in the serum under tumor conditions contribute to the downregulation of IFNAR. In summary, our results show that G-MDSCs initiates immunosuppressive functions in the peripheral
blood of tumor-bearing mice and that G-CSF participates in the acquisition of G-MDSC immunosuppressive functions by activating the PI3K-Akt/mTOR pathway. It is worth noting that the
downregulation of IFN-I signaling in G-MDSCs is a prerequisite for their immunosuppressive function. The downregulation of IFN-I signaling in G-MDSCs leads to the activation of the
PI3K-Akt/mTOR pathway by downregulating SOCS1 (Fig. 8). The results indicate that the generation of immunosuppressive G-MDSCs under tumor conditions involves a transformation from immune
activation to immune tolerance, providing a mechanistic insight for combating the immunosuppressive microenvironment. MATERIALS AND METHODS MICE AND CELL LINES C57BL/6J mice (8–10 weeks old)
were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). B16F10 cells (1 × 106) were implanted subcutaneously in female mice (_n_ = 4–6 mice/group).
Tumor growth was measured using calipers, and tumor volume was calculated as follows: _V_ = (length × width2) × 0.5. For all drug efficacy studies, Rapamycin and Stattic treatments (five
times per week) were initiated at 2 weeks of tumor-bearing. All mice were housed in specific pathogen-free conditions. All animal procedures and studies were conducted in accordance with the
Institutional Animal Care and Use Committee guidelines. Mouse melanoma B16F10 cells, MEF cells, human embryonic kidney HEK-293T and 32D clone 3 cells were purchased from American Type
Culture Collection. B16F10, MEF and HEK-293T cells were grown in DMEM supplemented with 10% heat-inactivated FBS (HyClone, Logan, UT, USA) and 1% penicillin/streptomycin. 32D clone 3 cells
were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS and 10% mouse interleukin IL-3 (213–13, Peprotech) culture supplement. All cells were routinely validated for
lack of Mycoplasma infection with LookOut Mycoplasma PCR Detection Kit (Sigma, Saint Louis, MO, USA) and used within 10 passages. REAGENTS AND ANTIBODIES Recombinant murine CXCL10 (250-16),
CXCL13 (250-24), G-CSF (250-05), and GM-CSF (315-03) were purchased from PeproTech (Rocky Hill, NJ, USA). Directly conjugated anti-mouse mAbs CD11b-FITC (M1/70), CD45-PE/Cy7 (30-F11),
Ly6G-APC/Cy7 (1A8), Ly6C-PE (AL-21) were from BD Pharmingen (San Jose, CA, USA). Carboxyfluorescein succinimidyl ester (CFSE) was from Molecular Probes (Eugene, OR, USA). The Annexin-V-APC
apoptosis analysis kit (AO2001-11P-G) was obtained from Sungene (Tianjin, China). The mouse cytokine array panel A (ARY006) was purchased from R&D Systems (Minneapolis, MN, USA). The
MDSC isolation kit was from Miltenyi Biotec (Bergisch Gladbach, Germany) and the streptavidin particles were from BD IMag (San Jose, CA, USA). Anti-p-Akt (Ser473, #4060), anti-Akt (#4691),
anti-p-mTOR (Ser2448, #5536), anti-mTOR (#2983), anti-p-STAT3 (Tyr705, #9145) Abs were purchased from Cell Signal Technology (Danvers, MA). Anti-STAT3 (F-2) Abs was purchased from Santa Cruz
Biotechnology (Santa Cruz, CA). Rapamycin (HY-10219) and Stattic (HY-13818) were purchased from Med Chem Express (New Jersey, USA). FLOW CYTOMETRY Single-cell suspensions from bone marrow
and peripheral blood were treated with hypotonic lysis buffer to eliminate erythrocytes, and stained with fluorochrome-conjugated antibodies at 4 °C for 30 min. Following tumor tissues
processing, single-cell suspensions were resuspended in FC blocked (CD16/32, BD #553141) on ice and then incubated with antibody master mix. Dead cells in tumor tissue were identified by
staining with 7-AAD (Biolegend) according to the manufacturer’s instructions. For apoptosis analysis, cells were stained with surface marker followed by Annexin-V staining in binding buffer
(Biolegend). The treated cells were tested by flow cytometry using a FACSCanto II (BD Biosciences, San Jose, CA, USA), and the data were analyzed with FACSDiva software (BD Biosciences) and
Flow Jo 7.6.1 software. CELL ISOLATION Bone marrow cells were isolated by flushing the femurs and tibias with PBS supplemented with 2% FBS. Splenocytes were prepared by grinding the spleen
against a 70-μm nylon mesh. CD8+ T cells were isolated from splenocytes using the Mouse CD8 T-cell Isolation Kit (Biolegend). To obtain single cell from tumors, tissues were minced and
subjected to 1 h enzymatic digestion using 0.5 mg/ml collagenase IV (Sigma) and 0.01 mg/ml DNase I (Sigma) in RPMI 1640 supplemented with 2% FBS at 37 °C. Single-cell suspensions were passed
through a 70-μm mesh followed by erythrocytes removal using ammonium chloride lysis buffer. For the isolation of G-MDSCs, cells were labeled with biotinylated antibody to Ly6G (Miltenyi
Biotec), incubated with streptravidin-coated microbeads (Miltenyi Biotec) and separated on MACS columns (Miltenyi Biotec). The purity and viability of the sorted cells is over 90%. IN VITRO
T-CELL PROLIFERATION ASSAY CD8+ T cells (2 × 105) from normal mice were pre-stained with 1 μM CFSE at room temperature for 5 min and then quenched with 10% FBS. The treated CD8+ T cells were
plated in U bottom 96-well plates in triplicates and stimulated with the antibodies of anti-CD3 (0.5 μg/ml) and anti-CD28 (1 μg/ml) in the presence of the same number of G-MDSCs. On day 3
of the coculture, cells were collected and evaluated by flow cytometry. RNA EXTRACTION AND Q-PCR Total RNA was extracted with TRIzol (Invitrogen), and the cDNA was synthesized with reverse
transcriptase (Thermo Fisher Scientific, Waltham, MA, USA). The cDNA was then used as template for semi-quantitative or quantitative PCR analysis with Power SYBR Green PCR Master Mix (Thermo
Fisher, Waltham, MA) using the StepOne Plus Real Time PCR System (Applied Biosystems, Foster City, CA). Q-PCR data were analyzed by the ΔΔCt method and relative expression of mRNA was
normalized to β-actin. The primer sequences used were listed in Supplementary Table 1. TUMOR METASTASIS ASSAY IN VIVO In total, 2 × 105 B16F10 cells were injected via the tail vein into
C57BL/6J mice and lung metastasis incidence was analyzed about 2 weeks later. To determinate the effects of G-MDSCs, C57BL/6J mice were intravenously injected with 2 × 105 B16F10 cells, and
followed immediately, 2 and 4 days later, by injection of 2 × 106 G-MDSC or mice were treated with intraperitoneal injections of 200 μg in 100 μl of rat IgG2b isotopic control or anti-GR1
antibody. CYTOKINE ARRAY AND ENZYME-LINKED IMMUNOSORBENT ASSAY (ELISA) OF SERUM Mouse peripheral blood samples were centrifuged at 3000 rpm for 10 min. Serum was collected, aliquoted, and
stored at −80 °C. Expression of cytokines in mouse serum was evaluated using mouse cytokine antibody array Panel A (ARY006, R&D Systems). The level of IFNα and VEGF in serum was
determined using ELISA kits (eBioscience) according to manufacturer’s instructions. CONSTRUCTION OF THE LENTIVIRAL EXPRESSION PLASMID AND GENE TRANSFECTION Synthesized short hairpin (sh)
RNAs sequences were cloned into pLVTHM vector, and the constructed plasmids or shCtrl plasmid were transfected into HEK-293T cells, together with the packaging plasmid psPAX2 and the
envelope plasmid pMD2.G (both from Addgene) using Lipofectamine 2000 reagent (Invitrogen). The collected supernatant was concentrated and intrapleurally injected into 2-week tumor-bearing
mice four times every other day. The relative sequences used were listed in Supplementary Table 2. RNA-SEQ ANALYSIS For RNA sequencing (RNA-seq), peripheral blood G-MDSCs was collected from
normal mice, mice bearing tumor for 2 and 3 weeks. The cellular RNA was extracted using Trizol reagent followed by a genomic DNA elimination step. RNA purity was assessed using the
kaiaoK5500® Spectrophotometer (Kaiao, Beijing, China). The RNA integrity and concentration was assessed using a RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies,
CA, USA). Library construction and sequencing on an Illumina HiSeq 2500 instrument was performed at Annoroad Gene Technology Corporation (Beijing, China). Bowtie2 v2.2.3 was used to build
the genome index, and clean data was then aligned to the reference genome using HISAT2 v2.1.0. The level of gene expression was quantified using a software package called FPKM (Fragments Per
Kilobase Millon Mapped Reads). DEGseq v1.18.0 was used for differential gene expression analysis. Overall changes were considered significant if they passed the FDR threshold of <5%. RNA
sequencing of the raw data and processed expression data for this study have been deposited in the NCBI Gene Expression Omnibus and are accessible through GEO Series accession number:
GSE141652. WESTERN BLOT Cells were harvested and solubilized in RIPA buffer containing protease and phosphatase inhibitors. 30 μg protein was electrophoresed on Biorad precast gradient gels
and electroblotted onto PVDF membranes. Proteins were detected by incubation with 1:1000 dilutions of primary antibodies, washed and incubated with Goat anti-rabbit-HRP antibodies or Goat
anti-mouse-HRP antibodies and detected after incubation with a chemiluminescent substrate. SIRNA-MEDIATED INTERFERENCE For RNA interference experiments, scramble control, or smart-pool siRNA
respectively against IFNAR and SOCS1 were transfected into 32D clone 3 cells using Lipofectamine 3000 according to the manufacturer’s instructions. siRNA duplex oligoribonucleotides were
synthesized by GenePharma (Shanghai, China). The sense sequences were as follows: si1-IFNAR, 5′-GUCUGAAAGUUUUGAUAGATT-3′; si2-IFNAR, 5′-CCGUUGUACUCUAGUACUUTT-3′; si1-SOCS1,
5′-CCAGUUUAGGUAAUAAACUTT-3′ and si2-SOCS1, 5′-ACACUCACUUCCGCACCUUTT-3′. CO-IMMUNOPRECIPITATION (CO-IP) ASSAY The cell lysates were extracted after transfection of flag-SOCS1 plasmid into 32D
cells for 30 h. Cell extracts were cleared with protein G beads for 1 h at 4 °C before being incubated with 2 μg of monoclonal anti-Akt or FLAG antibodies, or, alternatively, the same
amount of IgG, overnight at 4 °C. After washing, the precipitated proteins were analyzed by immunoblotting. GST PULL-DOWN ASSAY GST-Akt was cloned into the vectors pGEX-4T-2, and His-SOCS1
was cloned into pET-28a(+). _E. coli_ BL21 were transformed with pGEX-4T-2-Akt and pET-28a(+)-SOCS1 vectors. GST-Akt and His-SOCS1 protein expression were induced with IPTG and purified
using Glutathione Agarose 4B (Cytiva, #17075601) and Ni-NTA Agarose beads (Beyotime, #P2218) according to standard protocols. Purified GST-Akt and His-SOCS1 were incubated in reaction buffer
at 4 °C for 1 h, and then with GST-beads overnight at 4 °C. Afterwards, the beads were washed four times with low-salt buffer, and bound proteins were eluted and subjected to immunoblot
analysis. STATISTICS Statistical and graphical analyses were performed using Graphpad Prism. Results are presented as means ± SD. _t_-Test was performed using GraphPad Prism version 6
software. Differences were considered to be statistically significant when _p_ values were <0.05. All experiments were independently repeated at least three times. DATA AVAILABILITY All
study data are included in the article and/or supporting information. REFERENCES * Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. Article CAS
PubMed Google Scholar * Fox BA, Sanders KL, Chen S, Bzik DJ. Targeting tumors with nonreplicating Toxoplasma gondii uracil auxotroph vaccines. Trends Parasitol. 2013;29:431–7. Article CAS
PubMed PubMed Central Google Scholar * Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S. Myeloid-derived suppressor cells express the death receptor Fas
and apoptose in response to T cell-expressed FasL. Blood. 2011;117:5381–90. Article CAS PubMed PubMed Central Google Scholar * Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor
cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. Article CAS PubMed PubMed Central Google Scholar * Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor
cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506. Article CAS PubMed Google Scholar * Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat
Rev Cancer. 2013;13:739–52. Article CAS PubMed PubMed Central Google Scholar * Qu P, Wang LZ, Lin PC. Expansion and functions of myeloid-derived suppressor cells in the tumor
microenvironment. Cancer Lett. 2016;380:253–6. Article CAS PubMed Google Scholar * Tesi RJ. MDSC; the most important cell you have never heard of. Trends Pharm Sci. 2019;40:4–7. Article
CAS PubMed Google Scholar * Giallongo C, Romano A, Parrinello NL, La Cava P, Brundo MV, Bramanti V, et al. Mesenchymal stem cells (MSC) regulate activation of granulocyte-like myeloid
derived suppressor cells (G-MDSC) in chronic myeloid leukemia patients. PLoS ONE. 2016;11:e0158392. Article PubMed PubMed Central Google Scholar * Wang J, De Veirman K, Faict S,
Frassanito MA, Ribatti D, Vacca A, et al. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J Pathol.
2016;239:162–73. Article CAS PubMed Google Scholar * Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells
(MDSCs) during tumour progression. Br J Cancer. 2019;120:16–25. Article CAS PubMed Google Scholar * Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et
al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44. Article CAS PubMed Google
Scholar * Lang S, Bruderek K, Kaspar C, Hoing B, Kanaan O, Dominas N, et al. Clinical relevance and suppressive capacity of human myeloid-derived suppressor cell subsets. Clin Cancer Res.
2018;24:4834–44. Article CAS PubMed Google Scholar * Tian X, Tian J, Tang X, Rui K, Zhang Y, Ma J, et al. Particulate beta-glucan regulates the immunosuppression of granulocytic
myeloid-derived suppressor cells by inhibiting NFIA expression. Oncoimmunology. 2015;4:e1038687. Article PubMed PubMed Central Google Scholar * Zheng Y, Tian X, Wang T, Xia X, Cao F,
Tian J, et al. Long noncoding RNA Pvt1 regulates the immunosuppression activity of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Mol Cancer. 2019;18:61. Article
PubMed PubMed Central Google Scholar * Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–802.
Article CAS PubMed Google Scholar * Porritt RA, Hertzog PJ. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. 2015;36:150–60.
Article CAS PubMed Google Scholar * Lercher A, Bhattacharya A, Popa AM, Caldera M, Schlapansky MF, Baazim H, et al. Type I interferon signaling disrupts the hepatic urea cycle and alters
systemic metabolism to suppress T cell function. Immunity. 2019;51:1074–87. Article CAS PubMed PubMed Central Google Scholar * Parker BS, Rautela J, Hertzog PJ. Antitumour actions of
interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16:131–44. Article PubMed Google Scholar * Alicea-Torres K, Sanseviero E, Gui J, Chen J, Veglia F, Yu Q, et al. Immune
suppressive activity of myeloid-derived suppressor cells in cancer requires inactivation of the type I interferon pathway. Nat Commun. 2021;12:1717. Article CAS PubMed PubMed Central
Google Scholar * Snell LM, McGaha TL, Brooks DG. Type I interferon in chronic virus infection and cancer. Trends Immunol. 2017;38:542–57. Article CAS PubMed PubMed Central Google
Scholar * Osborn JL, Greer SF. Metastatic melanoma cells evade immune detection by silencing STAT1. Int J Mol Sci. 2015;16:4343–61. Article CAS PubMed PubMed Central Google Scholar *
Ortiz A, Gui J, Zahedi F, Yu P, Cho C, Bhattacharya S, et al. An interferon-driven oxysterol-based defense against tumor-derived extracellular vesicles. Cancer Cell. 2019;35:33–45. Article
CAS PubMed PubMed Central Google Scholar * Critchley-Thorne RJ, Simons DL, Yan N, Miyahira AK, Dirbas FM, Johnson DL, et al. Impaired interferon signaling is a common immune defect in
human cancer. Proc Natl Acad Sci USA. 2009;106:9010–5. Article CAS PubMed PubMed Central Google Scholar * Abrams SI, Waight JD. Identification of a G-CSF-Granulocytic MDSC axis that
promotes tumor progression. Oncoimmunology. 2012;1:550–1. Article PubMed PubMed Central Google Scholar * Salminen A, Kaarniranta K, Kauppinen A. The role of myeloid-derived suppressor
cells (MDSC) in the inflammaging process. Ageing Res Rev. 2018;48:1–10. Article CAS PubMed Google Scholar * Nam S, Kang K, Cha JS, Kim JW, Lee HG, Kim Y, et al. Interferon regulatory
factor 4 (IRF4) controls myeloid-derived suppressor cell (MDSC) differentiation and function. J Leukoc Biol. 2016;100:1273–84. Article CAS PubMed Google Scholar * Mackey JBG, Coffelt SB,
Carlin LM. Neutrophil maturity in cancer. Front Immunol. 2019;10:1912. Article CAS PubMed PubMed Central Google Scholar * Keeley T, Costanzo-Garvey DL, Cook LM. Unmasking the many
faces of tumor-associated neutrophils and macrophages: considerations for targeting innate immune cells in cancer. Trends Cancer. 2019;5:789–98. Article CAS PubMed Google Scholar *
Hawila E, Razon H, Wildbaum G, Blattner C, Sapir Y, Shaked Y, et al. CCR5 directs the mobilization of CD11b(+)Gr1(+)Ly6C(low) polymorphonuclear myeloid cells from the bone marrow to the
blood to support tumor development. Cell Rep. 2017;21:2212–22. Article CAS PubMed Google Scholar * Weichhart T, Saemann MD. The PI3K/Akt/mTOR pathway in innate immune cells: emerging
therapeutic applications. Ann Rheum Dis. 2008;67:iii70–4. Article CAS PubMed Google Scholar * Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, et al. The
TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–77. Article CAS PubMed Google Scholar * Jones LM, Broz ML, Ranger JJ, Ozcelik J, Ahn R, Zuo
D, et al. STAT3 establishes an immunosuppressive microenvironment during the early stages of breast carcinogenesis to promote tumor growth and metastasis. Cancer Res. 2016;76:1416–28.
Article CAS PubMed Google Scholar * Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer
patients. J Clin Invest. 2013;123:1580–9. Article CAS PubMed PubMed Central Google Scholar * Cherfils-Vicini J, Iltis C, Cervera L, Pisano S, Croce O, Sadouni N, et al. Cancer cells
induce immune escape via glycocalyx changes controlled by the telomeric protein TRF2. EMBO J. 2019;38:e100012. Article PubMed PubMed Central Google Scholar * Schoggins JW, Wilson SJ,
Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–5. Article CAS PubMed
PubMed Central Google Scholar * McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87–103. Article CAS PubMed PubMed
Central Google Scholar * Liu YF, Zhuang KH, Chen B, Li PW, Zhou X, Jiang H, et al. Expansion and activation of monocytic-myeloid-derived suppressor cell via STAT3/arginase-I signaling in
patients with ankylosing spondylitis. Arthritis Res Ther. 2018;20:168. Article PubMed PubMed Central Google Scholar * Qi X, Jiang H, Liu P, Xie N, Fu R, Wang H, et al. Increased
myeloid-derived suppressor cells in patients with myelodysplastic syndromes suppress CD8+ T lymphocyte function through the STAT3-ARG1 pathway. Leuk Lymphoma. 2021;62:218–23. Article CAS
PubMed Google Scholar * Guchhait P, Tosi MF, Smith CW, Chakaraborty A. The murine myeloid cell line 32Dcl3 as a model system for studying neutrophil functions. J Immunol Methods.
2003;283:195–204. Article CAS PubMed Google Scholar * Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, et al. Autophagy-dependent generation of free fatty acids is
critical for normal neutrophil differentiation. Immunity. 2017;47:466–80. Article CAS PubMed PubMed Central Google Scholar * Palmer DC, Restifo NP. Suppressors of cytokine signaling
(SOCS) in T cell differentiation, maturation, and function. Trends Immunol. 2009;30:592–602. Article CAS PubMed PubMed Central Google Scholar * Elliott J, Johnston JA. SOCS: role in
inflammation, allergy and homeostasis. Trends Immunol. 2004;25:434–40. Article CAS PubMed Google Scholar * Lopez-Sanz L, Bernal S, Recio C, Lazaro I, Oguiza A, Melgar A, et al.
SOCS1-targeted therapy ameliorates renal and vascular oxidative stress in diabetes via STAT1 and PI3K inhibition. Lab Invest. 2018;98:1276–90. Article CAS PubMed Google Scholar * Sugase
T, Takahashi T, Serada S, Fujimoto M, Ohkawara T, Hiramatsu K, et al. SOCS1 gene therapy has antitumor effects in imatinib-resistant gastrointestinal stromal tumor cells through FAK/PI3 K
signaling. Gastric Cancer. 2018;21:968–76. Article CAS PubMed Google Scholar * Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, et al. Structure and function of a new
STAT-induced STAT inhibitor. Nature. 1997;387:924–9. Article CAS PubMed Google Scholar * Ortiz ML, Kumar V, Martner A, Mony S, Donthireddy L, Condamine T, et al. Immature myeloid cells
directly contribute to skin tumor development by recruiting IL-17-producing CD4+ T cells. J Exp Med. 2015;212:351–67. Article CAS PubMed PubMed Central Google Scholar * Marvel D,
Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125:3356–64. Article PubMed PubMed Central Google Scholar *
Meyer C, Sevko A, Ramacher M, Bazhin AV, Falk CS, Osen W, et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse
melanoma model. Proc Natl Acad Sci USA. 2011;108:17111–6. Article CAS PubMed PubMed Central Google Scholar * Hsu BE, Tabaries S, Johnson RM, Andrzejewski S, Senecal J, Lehuede C, et al.
Immature low-density neutrophils exhibit metabolic flexibility that facilitates breast cancer liver metastasis. Cell Rep. 2019;27:3902–15. Article CAS PubMed Google Scholar * Kowanetz
M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci USA.
2010;107:21248–55. Article CAS PubMed PubMed Central Google Scholar * Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a
granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS ONE. 2011;6:e27690. Article CAS PubMed PubMed Central Google Scholar * Amano MT, Castoldi A, Andrade-Oliveira V,
Latancia MT, Terra FF, Correa-Costa M, et al. The lack of PI3Kgamma favors M1 macrophage polarization and does not prevent kidney diseases progression. Int Immunopharmacol. 2018;64:151–61.
Article CAS PubMed Google Scholar * Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature.
2016;539:437–42. Article CAS PubMed PubMed Central Google Scholar * Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–70. Article CAS PubMed Google Scholar *
Granot Z, Fridlender ZG. Plasticity beyond cancer cells and the “immunosuppressive switch”. Cancer Res. 2015;75:4441–5. Article CAS PubMed Google Scholar * Brandau S, Moses K, Lang S.
The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer: cousins, siblings or twins? Semin Cancer Biol. 2013;23:171–82. Article CAS PubMed Google Scholar *
Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, et al. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil
differentiation. J Biol Chem. 2004;279:44123–32. Article CAS PubMed Google Scholar * Taleb KA-O, Auffray C, Villefroy P, Pereira AA-O, Hosmalin AA-O, Gaudry M, et al. Chronic type I IFN
is sufficient to promote immunosuppression through accumulation of myeloid-derived suppressor cells. J Immunol. 2017;198:1156–63. Article CAS PubMed Google Scholar * Ma H, Yang W, Zhang
L, Liu S, Zhao M, Zhou G, et al. Interferon-alpha promotes immunosuppression through IFNAR1/STAT1 signalling in head and neck squamous cell carcinoma. Br J Cancer. 2019;120:317–30. Article
CAS PubMed Google Scholar * Zhang CX, Ye SB, Ni JJ, Cai TT, Liu YN, Huang DJ, et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell
expansion. Cell Death Differ. 2019;26:2314–28. Article CAS PubMed PubMed Central Google Scholar * Zheng H, Qian J, Carbone CJ, Leu NA, Baker DP, Fuchs SY. Vascular endothelial growth
factor-induced elimination of the type 1 interferon receptor is required for efficient angiogenesis. Blood. 2011;118:4003–6. Article CAS PubMed PubMed Central Google Scholar * Zheng H,
Qian J, Varghese B, Baker DP, Fuchs S. Ligand-stimulated downregulation of the alpha interferon receptor: role of protein kinase D2. Mol Cell Biol. 2011;31:710–20. Article CAS PubMed
Google Scholar * Bhattacharya S, Qian J, Tzimas C, Baker DP, Koumenis C, Diehl JA, et al. Role of p38 protein kinase in the ligand-independent ubiquitination and down-regulation of the
IFNAR1 chain of type I interferon receptor. J Biol Chem. 2011;286:22069–76. Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We would like to thank
Dr Huifang Shi (People’s Hospital of Rizhao) for many important discussions. This work was supported by grants from National Natural Science Foundation of China (Grant Nos. 31870888 and
81670095). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * The Key Laboratory of Molecular Epigenetics of Ministry of Education, Institute of Genetics and Cytology, School of Life Science,
Northeast Normal University, Changchun, Jilin, China Yingying Sun, Chao Shang, Yawei Wang, Boya Xu, Shu Jiang, Yan Mo, Dake Wang, Yueshuang Ke & Xianlu Zeng * Laboratory of Chemical
Biology, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin, China Xiaoqing Han Authors * Yingying Sun View author publications You can also search for
this author inPubMed Google Scholar * Xiaoqing Han View author publications You can also search for this author inPubMed Google Scholar * Chao Shang View author publications You can also
search for this author inPubMed Google Scholar * Yawei Wang View author publications You can also search for this author inPubMed Google Scholar * Boya Xu View author publications You can
also search for this author inPubMed Google Scholar * Shu Jiang View author publications You can also search for this author inPubMed Google Scholar * Yan Mo View author publications You can
also search for this author inPubMed Google Scholar * Dake Wang View author publications You can also search for this author inPubMed Google Scholar * Yueshuang Ke View author publications
You can also search for this author inPubMed Google Scholar * Xianlu Zeng View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Conception and
design: YS and XZ. Development of methodology: YS, XH, CS, and YW. Acquisition of data: YS, XH, CS, BX, SJ, and YM. Statistical analysis: YS and XH. Writing, review, and/or revision of the
manuscript: YS, CS, DW, YK, and XZ. CORRESPONDING AUTHORS Correspondence to Yueshuang Ke or Xianlu Zeng. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests.
ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by: Dr Lorenzo Galluzzi
SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE LEGENDS SUPPLEMENTARY FIG. 1 SUPPLEMENTARY FIG. 2 SUPPLEMENTARY FIG. 3 SUPPLEMENTARY FIG. 4 SUPPLEMENTARY FIG. 5 SUPPLEMENTARY FIG. 6
SUPPLEMENTARY FIG. 7 SUPPLEMENTARY FIG. 8 SUPPLEMENTARY FIG. 9 SUPPLEMENTARY TABLE 1 SUPPLEMENTARY TABLE 2 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative
Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the
original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in
the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended
use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sun, Y., Han, X., Shang, C. _et al._ The downregulation of type I IFN signaling in
G-MDSCs under tumor conditions promotes their development towards an immunosuppressive phenotype. _Cell Death Dis_ 13, 36 (2022). https://doi.org/10.1038/s41419-021-04487-w Download citation
* Received: 04 August 2021 * Revised: 01 December 2021 * Accepted: 20 December 2021 * Published: 10 January 2022 * DOI: https://doi.org/10.1038/s41419-021-04487-w SHARE THIS ARTICLE Anyone
you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the
Springer Nature SharedIt content-sharing initiative