dsDNA-induced AIM2 pyroptosis halts aberrant inflammation during rhabdomyolysis-induced acute kidney injury
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Rhabdomyolysis is a severe condition that commonly leads to acute kidney injury (AKI). While double-stranded DNA (dsDNA) released from injured muscle can be involved in its pathogenesis, the
exact mechanism of how dsDNA contributes to rhabdomyolysis-induced AKI (RIAKI) remains obscure. A dsDNA sensor, absent in melanoma 2 (AIM2), forms an inflammasome and induces gasdermin D
(GSDMD) cleavage resulting in inflammatory cell death known as pyroptosis. In this study using a mouse model of RIAKI, we found that Aim2-deficiency led to massive macrophage accumulation
resulting in delayed functional recovery and perpetuating fibrosis in the kidney. While Aim2-deficiency compromised RIAKI-induced kidney macrophage pyroptosis, it unexpectedly accelerated
aberrant inflammation as demonstrated by CXCR3+CD206+ macrophage accumulation and activation of TBK1-IRF3/NF-κB. Kidney macrophages with intact AIM2 underwent swift pyroptosis without IL-1β
release in response to dsDNA. On the other hand, dsDNA-induced Aim2-deficient macrophages escaped from swift pyroptotic elimination and instead engaged STING-TBK1-IRF3/NF-κB signalling,
leading to aggravated inflammatory phenotypes. Collectively, these findings shed light on a hitherto unknown immunoregulatory function of macrophage pyroptosis. dsDNA-induced rapid
macrophage cell death potentially serves as an anti-inflammatory program and determines the healing process of RIAKI.
Rhabdomyolysis is a pathological condition characterized by skeletal muscle breakdown, which elicits the release of intracellular contents into blood circulation. Kidneys are commonly
affected, and this manifests as rhabdomyolysis-induced acute kidney injury (AKI) (RIAKI). Patients with this life-threatening complication commonly require kidney replacement therapy, and
10–14% of patients die during hospitalization [1,2,3]. Moreover, recent evidence reveals that there is a significant risk that AKI will transition to chronic kidney disease (CKD) after a
single episode of RIAKI [3]. Intramuscular contents including danger-associated molecular patterns (DAMPs), such as myoglobin and heme, are considered to be nephrotoxic and contribute to the
onset of RIAKI. Recently, endogenously released double-stranded DNA (dsDNA) has also been suggested to play a role in its pathogenesis [4]. However, the precise mechanism by which dsDNA
contributes to kidney inflammation in this syndrome remains largely unknown.
The presence of dsDNA in the cytosol is recognized by a plethora of pattern recognition receptors (PRRs), leading to an innate immune response and/or cell death. Absent in melanoma 2 (AIM2)
is an inflammasome-forming DNA-sensing PRR [5, 6]. Inflammasomes are innate immune pathways that regulate inflammation and necrotic cell death called pyroptosis. AIM2 is comprised of one
N-terminal PYD domain and one C-terminal HIN domain, which preferentially binds to dsDNA in a non-sequence-specific manner. Upon activation, AIM2 assembles apoptosis-associated speck-like
protein containing a CARD (ASC) molecules via homotypic PYD-PYD interactions and then recruits pro-inflammatory caspase-1. Proximity-induced caspase-1 activation triggers the maturation of
interleukin (IL)-1β/IL-18 and the cleavage of gasdermin D (GSDMD), providing inflammation and pyroptosis. In CKD, the release of dsDNA from necrotic cells has been implicated in activation
of the AIM2 inflammasome [7]. Moreover, we previously reported that another inflammasome-forming PRR, NLRP3, is involved in the pathogenesis of RIAKI [8]. These observations led us to
hypothesize that the AIM2 inflammasome would recognize extracellular dsDNA during RIAKI and contribute to its consequences. On the other hand, numerous non-AIM2 DNA-sensing PRRs have been
identified to date. Most of them, such as cyclic GMP-AMP synthase (cGAS), induce the transcription of type I interferons (IFNs) and inflammation by activating the stimulator of interferon
genes (STING), TANK-binding kinase 1(TBK1), and interferon regulatory factor 3 (IRF3) pathway [9, 10]. It is currently unclear whether AIM2 and non-AIM2 DNA-sensing pathways interplay during
RIAKI.
In the present study, we examined the roles of dsDNA and AIM2 in a mouse model of RIAKI in wild-type (WT) and Aim2–/– mice. Unexpectedly, Aim2-deficiency did not influence the severity of
initial kidney injury, but did delay its recovery, and potentiated the subsequent inflammatory response and fibrosis. Loss of Aim2 limited macrophage pyroptosis and instead accelerated
subsequent inflammation by the recruitment of CXCR3+CD206+ macrophages during RIAKI. In in vitro experiments, dsDNA-treated Aim2-deficient macrophages engaged the STING-TBK1-IRF3/NF-κB
pathway and released pro-inflammatory cytokines. Accordingly, the lack of prompt elimination of inflammatory macrophages by pyroptosis led to aberrant inflammation and impaired normal
healing under Aim2-deficiency. Our results indicate that AIM2 pyroptosis is a critical mediator for the resolution of inflammation during RIAKI.
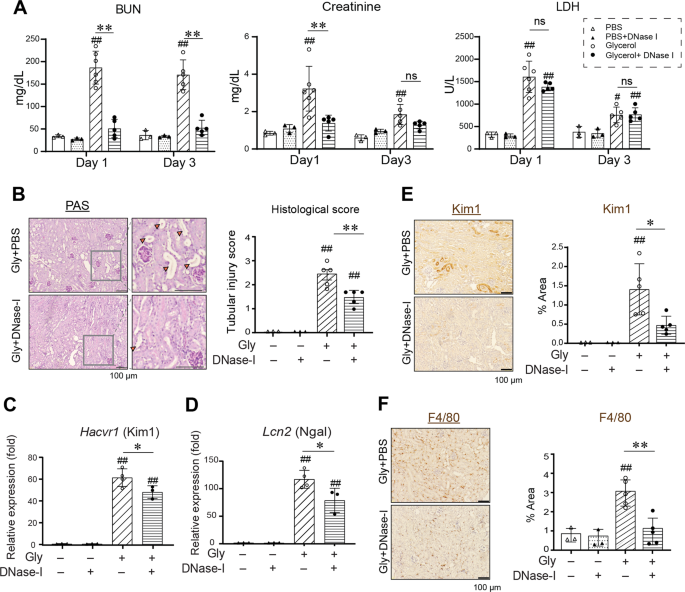
DNase-I is a potent endonuclease that hydrolyzes phosphodiester bonds and degrades extracellular double-stranded DNA (dsDNA) [11]. It reportedly mitigates the kidney dysfunction in RIAKI
[4]. To verify the role of dsDNA as a DAMP during RIAKI, we first re-analyzed the effects of DNase-I in a mouse RIAKI model. Intramuscular glycerol injection elicited kidney dysfunction as
characterized by increased serum BUN and creatinine levels, and DNase-I treatment significantly abrogated kidney dysfunction (Fig. 1A). This is not due to the effect of DNase-I on
rhabdomyolysis, since LDH release was comparable between groups given vehicle or DNase-I. DNase-I treatment alleviated histological tubular epithelial injury, the upregulation of kidney
injury molecule 1 (KIM1, or Hacvr1) and neutrophil gelatinase-associated lipocalin (Ngal, or Lcn2), and macrophage infiltration in the kidney after glycerol induction (Fig. 1B–F). These
results indicate that extracellular dsDNA is an essential danger molecule that contributes to both kidney tubular epithelial damage and the accumulation of immune cells during RIAKI.
DNase-I was administrated peritoneally just before and after glycerol injection and then every 12 h in WT mice. A Serum BUN, creatinine, and LDH levels at day 1 and day 3 (n = 3, 3, 6, 5 for
Day 1, and 3, 3, 5, 5 for Day 3). B PAS staining of the kidneys at day 3 after glycerol injection. Orange arrowheads indicate tubular epithelial loss, tubular dilatation, and cast
formation. Semi-quantitative assessment of tubular injury on PAS-stained sections (n = 3, 3, 5, and 5). C, D Kidney levels of Kim1 and Ngal mRNA were analyzed by real-time RT-PCR (n = 3, 3,
4, and 3). E Immunohistochemistry for the Kim1-positive area was quantified (n = 3, 3, 5, and 5). F Immunohistochemistry for the F4/80-positive area was quantified (n = 3, 3, 5, and 5). Data
are expressed as mean ± SD. *p