Carnosine suppresses human glioma cells under normoxic and hypoxic conditions partly via inhibiting glutamine metabolism
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
L-Carnosine (β-alanyl-L-histidine) is a naturally occurring dipeptide, which has shown broad-spectrum anticancer activity. But the anticancer mechanisms and regulators remain unknown. In
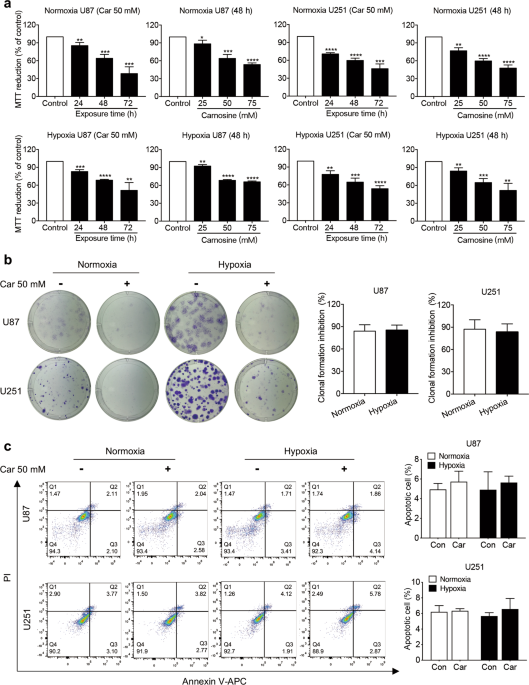
this study, we investigated the effects of carnosine on human glioma U87 and U251 cell lines under normoxia (21% O2) and hypoxia (1% O2). We showed that carnosine (25−75 mM) dose-dependently
inhibited the proliferation of the glioma cells; carnosine (50 mM) inhibited their colony formation, migration, and invasion capacity. But there was no significant difference in the
inhibitory effects of carnosine under normoxia and hypoxia. Treatment with carnosine (50 mM) significantly decreased the expression of glutamine synthetase (GS) at the translation level
rather than the transcription level in U87 and U251 cells, both under normoxia and hypoxia. Furthermore, the silencing of GS gene with shRNA and glutamine (Gln) deprivation significantly
suppressed the growth, migratory, and invasive potential of the glioma cells. The inhibitory effect of carnosine on U87 and U251 cells was partly achieved by inhibiting the Gln metabolism
pathway. Carnosine reduced the expression of GS in U87 and U251 cells by promoting the degradation of GS through the proteasome pathway, shortening the protein half-life, and reducing its
stability. Given that targeting tumor metabolism is a proven efficient therapeutic tactic, our results may present new treatment strategies and drugs for improving the prognosis of gliomas.
Gliomas, including astrocytoma, oligodendroglioma, and ependymoma, are tumors of neuroectoderm origin caused by glial cells or precursor cells [1]. Glioma is the most common type of
malignant brain tumor, accounting for 81% of malignant brain tumors, and the most lethal primary brain tumor [2, 3]. Its prognosis is poor because of the limited amount of tumor tissue that
can be safely removed, resistance of the residual tumor to radiotherapy and chemotherapy after operation, and blood–brain barrier, a challenge for drug delivery [4]. Glioma stem cells are
another very important feature of glioma. Glioma stem cells have strong DNA-repair mechanisms, leading to chemoradiotherapy resistance [5], and the ability to differentiate into stroma and
vascular structures that support tumor growth [6]. The need to find effective treatment methods and drugs for glioma is urgent.
Tumor hypoxia can strongly induce cells to develop aggressive and refractory phenotypes, leading to rapid progress and poor prognosis [7]. Hypoxia is a hallmark of gliomas, which, therefore,
histologically show the pathological characteristics of pseudopalisading necrosis and vascular hyperplasia [8]. On the other hand, a growing number of studies have found that mitochondrial
function in tumor cells is not absent but rather suppressed. Under some conditions, the metabolic activity of mitochondria in tumor cells is activated to promote the rapid growth of tumor
cells [9,10,11]. Both the glycolysis and mitochondrial oxidative phosphorylation pathways play important roles in tumor cell proliferation and/or metastasis. Therefore, the need to find new
tumor treatment strategies and drugs that can simultaneously target the glycolysis pathway and mitochondrial aerobic respiratory pathway is substantial.
Glutamine (Gln) was shown to play an important role in cell proliferation in the 1950s [12]. Gln synthesis is upregulated in some cancers; for example, some human gliomas accumulate large
amounts of Gln by synthesizing glucose-derived carbon under the catalysis of glutamine synthesis (GS) [13]. This promotes the de novo synthesis of purines and the biosynthesis of some
essential amino acids, making glioma cells self-sufficient in meeting the need for Gln. Consistent with this metabolic phenotype, GS is expressed in most gliomas [14]. Research has shown
that when primary glioblastoma cells maintain a stem cell-like state, GS expression is significantly increased, and glutamate (Glu) is taken up rather than released, rendering the growth of
glioblastoma-like cells independent of extracellular Gln [14].
Carnosine is a naturally occurring dipeptide composed of β-alanine and L-histidine [15]. In vivo and in vitro experiments have shown that carnosine is a powerful antioxidant [16], free
radical scavenger [17], and effective antiglycation agent [18]. In addition, carnosine is well tolerated in humans, has no known drug interactions or serious adverse reactions, and can
freely pass through the blood–brain barrier [19]. In recent years, the broad-spectrum antitumor effect of carnosine has attracted the attention of researchers [20, 21]. In our previous
studies on gastric cancer, we found that the antitumor effect of carnosine may be achieved by inhibiting both glycolysis and mitochondrial aerobic respiration [10, 22]. However, it is not
clear whether carnosine inhibits the proliferation, migration, and invasion of glioma cells by inhibiting glycolysis, mitochondrial aerobic metabolism, or both.
Therefore, in this study, we explored the effects of carnosine on the proliferation, migration, and invasion of glioma cells (U87 and U251 cells) under conditions of normoxia and hypoxia (1%
O2), and explored its potential molecular mechanism.
L-Carnosine was purchased from Sigma (St. Louis, MO, USA). Cycloheximide (CHX), chloroquine diphosphate (CLQ), and MG132 were purchased from MedChemExpress (Shanghai, China). A trypsin-EDTA
solution, BCA protein concentration detection kit, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) powder, immunostaining fixative, and TRIzol reagent were purchased
from Beyotime Institute of Biotechnology (Nanjing, China). Fetal bovine serum (FBS), high-glucose Dulbecco’s modified Eagle’s medium (DMEM), and Gln-free DMEM were obtained from GIBCO-BRL
(Grand Island, NY, USA). The PrimeScriptTM RT reagent kit and TB Green®Premix Ex TaqTM II were obtained from TakaRa Biotechnology Co., Ltd. (Dalian, China). An Annexin V-APC/PI apoptosis
detection kit was from KeyGenBiotech (Jiangsu, China).
The U87 human glioma cell line was purchased from the Institute of Cell Biology, Chinese Academy of Sciences (Shanghai). The U251 cell line was purchased from the China Center for Type
Culture Collection (CCTCC, Wuhan). Primary cultured rat cortical astrocytes were obtained through protocols described in our previously published article [23]. The cells were cultured in
DMEM (containing 4 mM Gln; the DMEM was used within 2 weeks to avoid Gln degradation) supplemented with 10% FBS, 100 U/mL penicillin G, and 100 μg/mL streptomycin. Some cells were cultured
at 37 °C in 5% CO2 and 21% O2 in a humidified incubator (Thermo Fisher Scientific, MA, USA) under normoxic conditions. Other cells were cultured at 37 °C in 5% CO2 and 1% O2 in a humidified
incubator (Galaxy 170R incubator, Eppendorf, Germany) under hypoxic conditions. Testing showed that the hypoxia performance of the tri-gas incubator was normal (Supplementary Fig. 1). Cells
in logarithmic growth phase were digested with trypsin at a ratio of 1:3. The subcultured cells were seeded onto 96- or 6-well plates at a density of 5 × 103 or 1 × 106 cells/well. For drug
treatments, 24 h after the cells were plated, they were treated with carnosine at different concentrations (25 mM, 50 mM, and 75 mM) for different intervals (24 h, 48 h, and 72 h). To
deplete Gln, cells were cultured in Gln-free DMEM.
Cell viability (mitochondrial activity) was detected by the MTT reduction assay. Cells were seeded onto 96-well plates at a density of 5 × 103 cells/well with three wells used for each
group. At the end of the experiment, the medium was discarded, and the cells were then incubated with 0.5 mg/mL MTT in a cell incubator for 4 h. Then, the supernatant was discarded, and 100
μL of DMSO was added to every well. After the crystal violet solution had completely dissolved, MTT metabolism was quantitated spectrophotometrically at 570 nm in a multimode microplate
reader (Thermo Fisher Scientific, MA, USA). The results are described as the percentage of MTT reduction, with the absorbance of the control group set at 100%.
Cells were seeded into six-well plates at a density of 250–300 cells per well with three wells used for each group and then treated with carnosine (50 mM). The cells were cultured for 14
days in DMEM supplemented with 1% FBS, 100 U/mL penicillin G, and 100 μg/mL streptomycin, and the medium was changed every 3 or 4 days. Finally, the cells were fixed with immunostaining
fixative and stained with a solution of crystal violet dye.
U87 and U251 cells were plated in 6-well plates at a density of 90%. The cells were scratched with a sterile 10-μL pipette tip. Then, the cells were washed with PBS to remove floating
cellular debris. The cells were then cultured in DMEM supplemented with 1% FBS and carnosine (50 mM). Every 12 h, the wounds were photographed, and the wound-closure rate was assessed by the
following formula: wound-closure rate (%) = (W0−Wt/W0) × 100%, where W0 is the wound width at 0 h and Wt is the wound width at a given time point (12–24 h).
Cell invasion assays were carried out using BD BioCoatMatrigel Invasion Chambers (24-well insert, 8-μm pore size, BD Biosciences, Bedford, MA) following the manufacturer’s instructions. U87
and U251 cells were trypsinized, resuspended in serum-free DMEM, and transferred to the upper chamber of Transwell inserts (5 × 104 cells/well). DMEM including 20% FBS was added to the lower
chamber. Cells were incubated for 24 h in a common cell incubator (21% O2) or hypoxia incubator (1% O2). Invasive cells were fixed with immunostaining fixative and stained with crystal
violet dye. Noninvasive cells in the inner part of the chambers were removed with cotton swabs. The invaded cells in five random fields were photographed with a microscope. Each experiment
was repeated three times. The cell migration assay was also performed with U87 cells and U251 cells with the procedure used for the invasion assay, except that the assay was performed
without the Matrigel inserts.
When cells in a dish had grown to ~80%–90% confluency, they were treated with carnosine (50 mM), CHX (150 μg/mL), the proteasome inhibitor MG132 (5 μM), and the lysosome inhibitor CLQ (25
μM). After treatment, the medium was removed, and the cells were washed with PBS twice. Cells were lysed on ice with RIPA lysate containing PMSF for 10 min, followed by centrifugation at
14,000 × g for 30 min at 4 °C. The supernatant was harvested in a new Eppendorf tube, and the protein concentration was quantified via BCA protein concentration detection kit. Western blot
analysis was performed by standard protocol. The following antibodies were used: mouse anti-tubulin (Beyotime Institute of Biotechnology, 1:1000), mouse anti-β-actin (Beyotime Institute of
Biotechnology, 1:1000), rabbit anti-GS (Abcam, 1:2000), mouse anti-HIF-1α (Abcam, 1:2000), HRP-labeled goat anti-rabbit IgG (1:1000), and HRP-labeled goat anti-mouse IgG (1:2000, Beyotime
Institute of Biotechnology, Nanjing, China).
To determine gene expression, total RNA was isolated with TRIzol reagent according to the manufacturer’s specifications, and purified RNA was quantified by spectrophotometry with a Nanodrop
(Thermo Fisher Scientific, MA, USA). cDNA was synthesized from 1 μg of total RNA using the PrimeScriptTM RT reagent kit according to the manufacturer’s guidelines. qRT-PCR to amplify GS was
performed using TB Green®Premix Ex TaqTM II with a CFX96 Real-Time PCR Detection System (Bio-Rad, CA, USA). The expression of β-actin was used as an internal control to normalize differences
in individual samples compared with the control sample. Target gene expression (relative mRNA expression) was calculated by the 2−ΔΔCt method, and is expressed as a fold change (mean ± SD)
over the average β-actin expression, which was set at one. The qRT-PCR primers used for the experiment were as follows: GS (Fw: 5′-TCATCTTGCATCGTGTGTGTG-3′; Rev: 5′-CTTCAGACCATTCTCCTCCCG-3′)
and β-actin (Fw: 5′-CCCTGGCACCCAGCAC-3′; Rev: 5′-GCCGATCCACACGGAGTAC-3′).
Cells were stained with Annexin V-APC and PI to evaluate apoptosis by flow cytometry according to the manufacturer’s instructions (KeyGenBiotech, Jiangsu, China). Briefly, 1 × 106
trypsinized cells were washed with PBS and stained with 5 μL of PI and 5 μL of Annexin V-APC in 500 μL of 1 × binding buffer for 5 min at room temperature in the dark. Quantification of
apoptotic cells was performed with a CytoFLEX (Beckman Coulter, CA, USA).
Three different small-hairpin RNAs (shRNAs) targeting GS and a negative control (NC) sequence were inserted into the GV112 vector (GeneChem, Shanghai, China). After verification by gene
sequencing, GV112-shLenti, the relevant NC plasmid, and two packaging plasmids (psPAX2 and pMD2.G, donated by Dr. Hong-zhi Li, Wenzhou Medical University, Wenzhou, China) were transfected
into 293T cells for 48 h using PolyJet (SignaGen, MD, USA). U87 and U251 cells were seeded in 60-mm dishes and cultured in DMEM for 24 h. Then, solution containing the virus was harvested,
filtered, and used to infect the cell lines with polybrene (8 μg/mL). The expression level of GS in transfected cells was confirmed by real-time PCR and Western blotting. The cell line with
the highest knockout efficiency was selected for follow-up experiments. The shRNA sequences used were as follows:
All data are presented as the mean ± SD of three or more independent experiments. Statistical analyses were conducted by SPSS 20.0. One-way ANOVA (analysis of variance) was applied for
multiple comparisons, whereas comparisons between two groups were analyzed using Student’s t test. P