Utmost, a single and cross-tissue twas (transcriptome wide association study), reveals new asd (autism spectrum disorder) associated genes
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Autism spectrum disorders (ASD) is a complex neurodevelopmental disorder that may significantly impact on the affected individual’s life. Common variation (SNPs) could explain about
50% of ASD heritability. Despite this fact and the large size of the last GWAS meta-analysis, it is believed that hundreds of risk genes in ASD have yet to be discovered. New tools, such as
TWAS (Transcriptome Wide Association Studies) which integrate tissue expression and genetic data, are a great approach to identify new ASD susceptibility genes. The main goal of this study
is to use UTMOST with the publicly available summary statistics from the largest ASD GWAS meta-analysis as genetic input. In addition, an in silico biological characterization for the novel
associated loci was performed. Our results have shown the association of 4 genes at the brain level (_CIPC, PINX1, NKX2-2_, and _PTPRE_) and have highlighted the association of _NKX2-2,
MANBA, ERI1,_ and _MITF_ at the gastrointestinal level. The gastrointestinal associations are quite relevant given the well-established but unexplored relationship between ASD and
gastrointestinal symptoms. Cross-tissue analysis has shown the association of _NKX2-2_ and _BLK_. UTMOST-associated genes together with their in silico biological characterization seems to
point to different biological mechanisms underlying ASD etiology. Thus, it would not be restricted to brain tissue and it will involve the participation of other body tissues such as the
gastrointestinal. SIMILAR CONTENT BEING VIEWED BY OTHERS COMMON GENETIC RISK VARIANTS IDENTIFIED IN THE SPARK COHORT SUPPORT DDHD2 AS A CANDIDATE RISK GENE FOR AUTISM Article Open access 03
August 2020 SYSTEMATIC ANALYSES OF GWAS SUMMARY STATISTICS FROM UK BIOBANK IDENTIFIED NOVEL SUSCEPTIBILITY LOCI AND GENES FOR UPPER GASTROINTESTINAL DISEASES Article 18 May 2023 A HUMAN
LEUKOCYTE ANTIGEN IMPUTATION STUDY UNCOVERS POSSIBLE GENETIC INTERPLAY BETWEEN GUT INFLAMMATORY PROCESSES AND AUTISM SPECTRUM DISORDERS Article Open access 06 July 2023 INTRODUCTION Autism
spectrum disorders (ASD) includes a range of neurodevelopmental disorders (NDDs) with onset in early development that are characterized by deficits in communication and social interactions,
as well as by repetitive patterns of behavior and restrictive interests1. ASD is a complex genetic disorder, involving both environmental and genetic factors. Although an important part of
the genetic architecture of ASD is unknown, it is considered that thousands of genes may be involved even most of them remain unidentified and functionally uncharacterized. Rare genetic
variation only explains 3% of ASD genetic risk even if it confers a high individual risk2. However, common variation (SNPs; _single nucleotide polymorphisms_) could explain about 50% of ASD
heritability. The most recent and the largest ASD GWAS meta-analysis done until now, including 18,381 ASD cases and 27,969 controls, has reported 93 genome-wide significant markers in three
separate loci (top SNP: rs910805; _p_-value: 2.04 × 10−9)3. Another methodological approach for common variation are gene-based association analysis (GBA) methods that employ the _p_-values
for each SNP within a gene to obtain a single statistic at this level. Thus, MAGMA has identified 15 genes, most of them located near the genome-wide significant SNPs identified in GWAS, but
7 genes have revealed association in four additional loci (_KCNN2_, _MMP12, NTM,_ and a cluster of genes on chromosome 17)3. Additional GBA methods using other algorithms as PASCAL have
helped to define the association of other genes located in the same LD region than those found by MAGMA (_NKX2-4_, _NKX2-2_, _CRHR1-IT1_, _C8orf74_, and _LOC644172_)4. In addition to GBA,
bioinformatic approaches that integrate functional data are increasingly used to highlight new genes underlying GWAS summary statistics. Transcriptome-wide association studies (TWAS) have
emerged as useful tools to study the genetic architecture of complex traits. Among them, MetaXcan5 and FUSION6 are well-known TWAS methods. UTMOST (unified test for molecular signatures) has
been recently reported as a novel framework for single and cross-tissue expression imputation. UTMOST is able to consider the joint effect of SNPs (summary statistics) across LD regions
(1000 Genomes) and to integrate tissue expression data (GTeX) creating single and cross-tissue covariance matrices that will help to define the gene-trait associations. UTMOST performance
was demonstrated at several levels and its accuracy was also well proved as it was able to identify a greater number of associations in biologically relevant tissues for complex diseases7.
The main aim of this paper is to further mine the summary data from the largest ASD meta-analysis using UTMOST. In addition, an in silico biological characterization for the novel associated
loci will be carried out using bioinformatic approaches (DEG, pathway, gene network, and an exploratory enhancer analysis). Overall, our results have demonstrated the association of _CIPC,
PINX1, NKX2-2,_ and _PTPRE_ at the brain level and have also revealed the relevance of gastrointestinal tissue in ASD etiology through the association of other genes (_NKX2-2, MANBA, ERI1,_
and _MITF)_. MATERIALS AND METHODS DATASETS Summary statistics from the latest ASD GWAS meta-analysis were obtained from the public repository available in the PGC website
(http://www.med.unc.edu/pgc/results-and-downloads). The following data set was employed: iPSYCH_PGC_ASD_Nov2017.gz (Grove et al.3) which includes the meta-analysis of ASD by the Lundbeck
Foundation Initiative for Integrative Psychiatric Research (iPSYCH) and the Psychiatric Genomics Consortium (PGC) released in November 2017. The data set comprises a total of 18,381 cases
and 27,969 controls. Additional information about the genotyping, QC methods, and Ethics Commitees as well as informed consents employed in are available at the PGC website and in the
previous Grove et al.3 study. TWAS ANALYSIS USING UTMOST UTMOST7 (https://github.com/Joker-Jerome/UTMOST) was run as a single tissue association test for 44 GTeX tissues
(single_tissue_association_test.py) and a cross tissue association test combining gene-trait associations was run by the joint GBJ test (joint_GBJ_test.py). Both tests use the previous
summary statistics of the ASD GWAS meta-analysis as an input3. UTMOST pre-calculated covariance matrices for single-tissue (covariance_tissue/) and joint test (covariance_joint/) were
downloaded. Other necessary command parameters were used by default. Transcriptome-wide significance for single tissue analysis was established as _p_-value = 3.85 × 10−6 (0.05/12984(maximum
number of genes tested) for brain tissues and _p_-value = 3.42 × 10−6 (0.05/14586 (maximum number of genes tested) for non-brain tissues after Bonferroni correction. Transcriptome-wide
significance for joint test was established as _p_-value = 3.27 × 10−6 (0.05/15274) considering the number of effective test (Tables 1–3) (Supplementary.csv files for each tissue).
Covariances matrices are only available for the 44 tissues of GteX v6: adipose subcutaneous, adipose visceral omentum, adrenal gland, artery aorta, artery coronary, artery tibial, brain
anterior cingulate cortex BA24, brain caudate basal ganglia, brain cerebellar hemisphere, brain cerebellum, brain cortex, brain frontal cortex BA9, brain hippocampus, brain hypothalamus,
brain nucleus accumbens basal ganglia, brain putamen basal ganglia, breast mammary tissue, cells EBV-transformed lymphocytes, cells transformed fibroblasts, colon sigmoid, colon transverse,
esophagus gastroesophageal junction, esophagus mucosa, esophagus muscularis, heart atrial appendage, heart left ventricle, liver, lung, muscle skeletal, nerve tibial, ovary, pancreas,
pituitary, prostate, skin not sun-exposed suprapubic, skin sun-exposed lower leg, small intestine, terminal ileum, spleen, stomach, testis, thyroid, uterus, vagina, whole blood. Morpheus
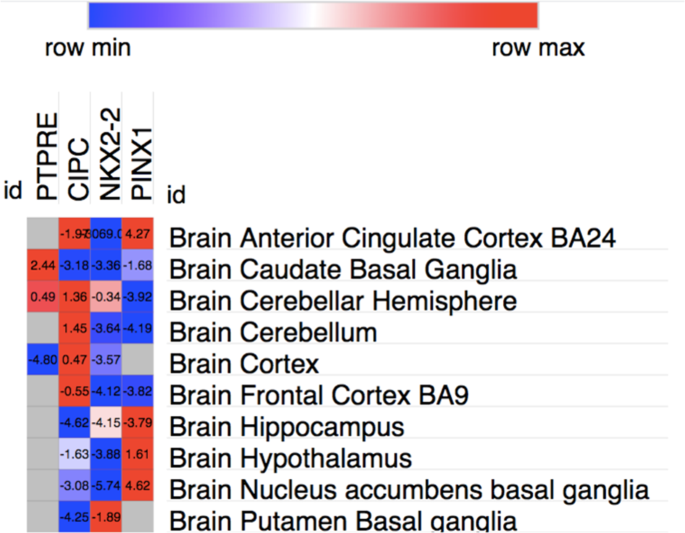
software (https://software.broadinstitute.org/morpheus/) was used to display the _Z_ scores of UTMOST significant genes across GTeX Brain tissues. UTMOST significance as a _Z_ score in brain
tissues is ∼4.6. Gray squares in the heatmap indicate that the gene weights were not available in the target tissue (Fig. 1). DEG AND GENE EXPRESSION ANALYSIS WITH FUMA GENE2FUNC, a tool of
FUMA8 (https://fuma.ctglab.nl/), was employed to carry out a gene expression heatmap and an enrichment analysis of differentially brain expressed genes (DEG) using BrainSpan RNA-seq data.
Those genes represented in Table 4 (bold: _PTPRE, CIPC, NKX2-2, PINX1_) were used as an input. Expression values are TPM (Transcripts Per Million) for GTEx v6 and RPKM (Read Per Kilobase per
Million). In order to define DEG sets, two-sided Student’s _t_-test were performed for these genes and per tissue against the different tissue types or developmental stages. Those genes
with a _p_-value < 0.05 after Bonferroni correction and a log fold change ≥0.58 are defined as DEG. The direction of expression was considered. The −log10 (p-value) refers to the
probability of the hypergeometric test DEG analysis was carried out creating differentially expressed genes for each expression data set (Fig. 2). Heatmaps display the normalized expression
value (zero mean normalization of log2 transformed expression), and darker red means higher relative expression of that gene in each label, compared to a darker blue color in the same label
(Fig. 3). GENEMANIA AND METASCAPE ANALYSIS GeneMANIA9 (https://genemania.org/) was used to build a gene network for the UTMOST-associated genes by the single tissue analysis (brain and
gastrointestinal tissues) and by the joint tissue analysis (Table 4). Each gene-network was subsequently analyzed with Metascape (https://metascape.org/)10 to carry out a pathway enrichment
and a protein–protein interaction enrichment using the Evidence Counting (GPEC) prioritization tool. For each given gene list, pathway and process enrichment analysis has been carried out
with the following ontology sources: GO biological processes, GO cellular components and GO molecular functions. The enrichment background includes all the genes in the genome. Terms with a
_p_-value < 0.01, a minimum count of 3, and an enrichment factor >1.5 (the enrichment factor is the ratio between the observed counts and the counts expected by chance) are collected
and grouped into clusters based on their membership similarities. More specifically, _p_-values are calculated based on the accumulative hypergeometric distribution, and _q_-values are
calculated using the Benjamini–Hochberg procedure to account for multiple testings. Kappa scores are used as the similarity metric when performing hierarchical clustering on the enriched
terms, and sub-trees with a similarity of >0.3 are considered a cluster. We select the terms with the best _p_-values from each of the 20 clusters, with the constraint that there are no
more than 15 terms per cluster and no more than 250 terms in total (Figs. 4–6). The network is visualized using Cytoscape, where each node represents an enriched term and is colored first by
its cluster ID (or each given gene list, protein–protein interaction enrichment analysis has been carried out with the following databases: BioGrid6, InWeb_IM7, OmniPath8. The resultant
network contains the subset of proteins that form physical interactions with at least one other member in the list. If the network contains between 3 and 500 proteins, the Molecular Complex
Detection (MCODE) algorithm has been applied to identify densely connected network components. The MCODE networks identified for individual gene lists are shown in Figs. 4–6. ENHANCER
ANALYSIS (DBSUPER) dbSUPER (https://asntech.org/dbsuper/) was used to perform an exploratory enhancer analysis for the UTMOST-associated genes (single-tissue analysis) and for those tissues
(brain and gastrointestinal) available at dbSUPER. The parameters selected were: SEs ranking method (H3K27ac), the peak calling was done with MACS (version 1.4.1) with parameters -p
1e-9,-keep-dup = auto, -w -S -space = 50, the stitching distance was established at 12,5 kb, the TSS exclusive zone was set at ±2 kb and the enhancer gene assignment was done within a 50 kb
window. RESULTS AND DISCUSSION UTMOST ANALYSES AND COMPARISON WITH PREVIOUS RESULTS UTMOST single tissue analysis (Brain tissues) showed association of two loci, _NKX2-2_ and _PTPRE_, while
other two loci, _CIPC_ and _PINX1_, showed a marginal association according to Bonferroni threshold (Table 1, Supplementary.csv files). _NKX2-2_ was previously identified as an associated
gene by two different GBA algorithms, MAGMA, and PASCAL3,4. The results obtained by UTMOST also serve to indicate the nucleus accumbens basal ganglia as the brain area in which these genes
may be acting. Although the association of _PTPRE_ was not obtained as such in previous analyses, the association of its neighboring gene, _C8orf74_, was noted in a previous study and one of
the index SNPs in the latest ASD GWAS was located near _PINX1_ (rs10099100) (Supplementary Fig. 1a, b)3,4. _NKX2-2_ was also marginally associated by UTMOST within non-brain tissue together
with other genes. It should be noted that some genes are tissue-specific for gastric and intestinal tissues such as stomach, esophagus and colon (_MANBA, ERI1, MITF,_ and _NKX2-2_). The
association of _NKX2-2_ in colon is noteworthy because _NKX2-2_ was previously reported as an ASD risk gene and now it is highlighted again by UTMOST in brain tissues. It seems that _NKX2-2_
together with _BLK_ may play a role in ASD etiology but not only at the brain level since UTMOST cross-tissue analysis also found association for both genes (Tables 2, 3, Supplementary
Figs. 1a, 2a, b, Supplementary.csv files) To evaluate the importance of each brain tissue in ASD etiology, a secondary analysis was performed using _Z_ scores values for each ASD-associated
gene across GTeX brain tissues. The heatmap showed that there is a wide specificity of association in terms of _Z_ score for each gene and brain tissue indicating the importance of
conducting tissue specific analyses such as UTMOST (Fig. 1). DEG ANALYSIS WITH GENE2FUNC TOOL (FUMA) DEG Analysis with BrainSpan data (11 general developmental stages of brain samples and 29
different ages of brain samples) for the brain associated set of genes have not shown any significant result (Fig. 2a, b). However, _CIPC_ have shown overexpression across every single
developmental stage in comparison with the remaining ASD-associated genes (Fig. 3a, b). GENEMANIA AND METASCAPE ANALYSIS GeneMANIA was used to find out the possible interactors with the
associated genes (Table 4). We have proposed three different analyses. One based on the genes associated in brain tissues; another one based on the associated genes at a gastrointestinal
level, given the associations pointed out by UTMOST and the previous implications of gastrointestinal abnormalities and symptoms in ASD and the lack of biological knowledge about them. The
final analysis is focused on the genes identified by the cross-tissue analysis and their interactors. The general goal is to delineate the biological pathways underlying each group of genes
and the differences between them. Gene network, enriched ontology clusters, and PPI interaction analysis for the brain-associated genes (_CIPC, PINX1, NKX2-2_, and _PTPRE_) by UTMOST Single
tissue analysis and their interactors highlights different biological pathways mainly involved in telomere maintenance and transcription regulation (Tables 5, 6 and Fig. 4). However,
associated genes in gastrointestinal tissue (_NKX2-2, MANBA, ERI1_, and _MITF_) and their interactors mainly regulate DNA transcription by RNA polymerase II and the fate of oligodendrocytes.
This is an interesting finding given the possible involvement of these cells in the enteric nervous system in ASD (Tables 7, 8 and Fig. 5). Finally, _NKX2-2_ and _BLK_ both associated in
the cross-tissue analysis, seem to point to very diverse biological pathways such as protein tyrosine kinase activity, B cell receptor complex, regulation of DNA-binding transcription factor
activity, and neuron projection morphogenesis, among others (Tables 8–10 and Fig. 6). ENHANCER ANALYSIS (DBSUPER) Given the tissue specificity given by UTMOST associations, we found
interesting to perform an exploratory analysis of enhancers. According to the dbSUPER dabase only _CIPC_ could work as a superenhancer in the brain middle hippocampus (SE_06106; chr14:
77562660-77607123, size: 44463 pb) (Supplementary Fig. 3). As far as we know, this is the first study that has employed the UTMOST framework combined with the summary statistics of the
largest ASD meta-analysis. The main aim was to identify ASD tissue-specific genes in brain and/or other tissues. It should be noted at the outset that gene-level associations identified by
UTMOST do not imply causality. However, looking at the regional plots for each loci associated by UTMOST, the results shown at brain and gastrointestinal level seems pretty consistent. Thus,
UTMOST has served to identify new ASD-associated genes in brain tissues as _PTPRE_ and _CIPC_. Furthermore, UTMOST has been useful to confirm and obtain information on the tissue in which
other known ASD risk genes such as _NKX2-2_ and P_INX1_ may have functional relevance. Altogether, brain-associated genes seems to point to three brain areas: cortex, hippocampus and nucleo
accumbens. Previous studies demonstrated the ASD-associated gene expression in brain cortex11. In addition, hippocampus underlie some of the featured social memory and cognitive behaviors
both crucial aspects in ASD12. The nucleus accumbens is a key brain area also related with the social reward response in ASD13. It should be also noted some limitations of our study. UTMOST
uses GTeX v6 data by default and it should be interesting to re-run the analysis once the tool will be updated. Thus, UTMOST could find novel associated genes when expression data from other
relevant ASD brain tissues as the amygdala are included. Another limitation of our results is the small number of SNPs considered by UTMOST to create the statistic in some genes (_PTPRE_ in
brain tissues and _NKX2-2_ in non-brain tissues analysis). However, we have been very conservative in establishing Bonferroni correction for all tissues in an analysis group. Thus, for
example, we always chose the maximum number of genes tested in one of the brain tissues and applied this to calcule Bonferroni for the whole group of brain tissues as a whole. In relation to
the biological pathways in which UTMOST-associated genes are involved, our results open possible avenues for future genetic and functional studies. Thus, the functional role in ASD of
_CIPC, PINX1, NKX2-2_, and _PTPRE_ has not yet been characterized in detail. Metascape analysis have provided an insight revealing their involvement in telomerase maintenance and
transcription regulation. Thus, _PINX1_ (_PIN2 (TERF1) Interacting Telomerase Inhibitor 1_) enhances TRF1 binding to telomeres and inhibits telomerase activity. It was proved that its
silencing compromises telomere length maintenance in cancer cells14. In addition, it was recently found that children with ASD and sensory symptoms have shorter telomeres, compared to those
children exhibiting a typical development15. _CIPC_ (_CLOCK interacting pacemaker_) is a mammalian circadian clock protein. Recently, circadian rithms were pointed out as involved in brain
development and they could underly ASD etiology due to its implication in behavioral processes. Circadian rythms are regulated through several transcription factors in different cellular
types, fact that might be related with the GO terms associated with transcription regulation in this study16. Moreover, the result of _CIPC_ as a predicted superenhancer highlights its
possible functional repercussion. UTMOST found association of several genes in non-brain tissues. However, we found really interesting to study those genes related to gastrointestinal
tissues (_NKX2-2, MANBA, ERI1_, and _MITF_). There is a well-known and established comorbidity among gastrointestinal symptoms and ASD17. These clinical associations suggest the implication
of gastrointestinal populations of neuronal cells. A mutation in _NLGN3_ (R451C) has been recently identified in two ASD brothers with GI symptoms. Mice models have demonstrated that R451C
alter the number of neuronal cells in the small intestine and impact fecal microbes18. These evidence suggest that the role of _NKX2-2, MANBA, ERI1_, and _MITF_ in gastrointestinal tissue
should be further studied. _BLK_ and _NKX2-2_ are both associated in the cross-tissue analysis. The importance of UTMOST approach is that is able to show the association of two previously
known ASD risk genes but not restricted to brain tissue. These findings lead to a difficult question whether autism can be a multisystemic disorder, something that has been recently pointed
out by some authors19. In conclusion, UTMOST, a novel single and cross-tissue TWAS, has revealed new ASD-associated genes. These genes have been characterized at the pathway and gene network
level using bioinformatic approaches. However, future tissue-specific functional studies will be key to properly determine their role in ASD etiology. REFERENCES * American Psychiatric
Association_. Diagnostic and Statistical Manual of Mental Disorders_ (American Psychiatric Publishing) (2013). * Sanders, S. J. et al. Insights into autism spectrum disorder genomic
architecture and biology from 71 risk loci. _Neuron_ 87, 1215–1233 (2015). Article CAS Google Scholar * Grove, J. et al. Identification of common genetic risk variants for autism spectrum
disorder. _Nat. Genet._ 51, 431–444 (2019). Article CAS Google Scholar * Alonso-Gonzalez, A., Calaza, M., Rodriguez-Fontenla, C. & Carracedo, A. Novel gene-based analysis of ASD
GWAS: insight into the biological role of associated genes. _Front. Genet._ 10, 733 (2019). Article CAS Google Scholar * Barbeira, A. N. et al. Exploring the phenotypic consequences of
tissue specific gene expression variation inferred from GWAS summary statistics. _Nat. Commun._ 9, 1825 (2018). Article Google Scholar * Gusev, A. et al. Integrative approaches for
large-scale transcriptome-wide association studies. _Nat. Genet._ 48, 245–252 (2016). Article CAS Google Scholar * Hu, Y. et al. A statistical framework for cross-tissue
transcriptome-wide association analysis. _Nat. Genet._ 51, 568–576 (2019). Article CAS Google Scholar * Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping
and annotation of genetic associations with FUMA. _Nat. Commun._ 8, 1826 (2017). Article Google Scholar * Warde-Farley, D. et al. The GeneMANIA prediction server: biological network
integration for gene prioritization and predicting gene function. _Nucleic Acids Res._ 38, W214–220 (2010). Article CAS Google Scholar * Zhou, Y. et al. Metascape provides a
biologist-oriented resource for the analysis of systems-level datasets. _Nat. Commun._ 10, 1523 (2019). Article Google Scholar * Voineagu, I. et al. Transcriptomic analysis of autistic
brain reveals convergent molecular pathology. _Nature_ 474, 380–384 (2011). Article CAS Google Scholar * Hitti, F. L. & Siegelbaum, S. A. The hippocampal CA2 region is essential for
social memory. _Nature_ 508, 88–92 (2014). Article CAS Google Scholar * Park, H. R. et al. A short review on the current understanding of autism spectrum disorders. _Exp. Neurobiol._ 25,
1–13 (2016). Article Google Scholar * Zhang, B. et al. Silencing PinX1 compromises telomere length maintenance as well as tumorigenicity in telomerase-positive human cancer cells. _Cancer
Res._ 69, 75–83 (2009). Article CAS Google Scholar * Lewis, C. R. et al. Telomere length and autism spectrum disorder within the family: relationships with cognition and sensory symptoms.
_Autism Res_. 13, 1094–1101 (2020). * Geoffray, M.-M., Nicolas, A., Speranza, M. & Georgieff, N. Are circadian rhythms new pathways to understand Autism Spectrum Disorder? _J. Physiol.
Paris_ 110, 434–438 (2016). Article Google Scholar * Buie, T. et al. Evaluation, diagnosis, and treatment of gastrointestinal disorders in individuals with ASDs: a consensus report.
_Pediatrics_ 125, S1–18 (2010). Article Google Scholar * Hosie, S. et al. Gastrointestinal dysfunction in patients and mice expressing the autism‐associated R451C mutation in neuroligin‐3.
_Autism Res._ 12, 1043–1056 (2019). Article Google Scholar * Thom, R. P. et al. Beyond the brain: A multi-system inflammatory subtype of autism spectrum disorder. _Psychopharmacol.
(Berl.)_ 236, 3045–3061 (2019). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank the Psychiatric Genomics Consortium Autism Spectrum Disorder Working Group for
making the ASD genome-wide association study results publicly available. Instituto de Salud Carlos III (ISCIII)/PI1900809/Cofinanciado FEDER. AUTHOR INFORMATION Author notes * These authors
contributed equally: Cristina Rodriguez-Fontenla, Angel Carracedo AUTHORS AND AFFILIATIONS * Grupo de Medicina Xenómica, Center for Research in Molecular Medicine and Chronic Diseases
(CiMUS), Universidad de Santiago de Compostela, Santiago de Compostela, Spain Cristina Rodriguez-Fontenla & Angel Carracedo * Fundación Pública Galega de Medicina Xenómica (FPGMX),
Centro de Investigación Biomédica en Red, Enfermedades Raras (CIBERER), Universidad de Santiago de Compostela, Santiago de Compostela, Spain Angel Carracedo Authors * Cristina
Rodriguez-Fontenla View author publications You can also search for this author inPubMed Google Scholar * Angel Carracedo View author publications You can also search for this author
inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Cristina Rodriguez-Fontenla. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare no competing interests. ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
FIGURES SUPPLEMENTARY FILES RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a
credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted
use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Rodriguez-Fontenla, C., Carracedo, A. UTMOST, a single and cross-tissue TWAS (Transcriptome Wide Association Study), reveals new ASD (Autism Spectrum Disorder)
associated genes. _Transl Psychiatry_ 11, 256 (2021). https://doi.org/10.1038/s41398-021-01378-8 Download citation * Received: 25 September 2020 * Revised: 22 March 2021 * Accepted: 08
April 2021 * Published: 30 April 2021 * DOI: https://doi.org/10.1038/s41398-021-01378-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative