Discovery of a novel egfr ligand dpba that degrades egfr and suppresses egfr-positive nsclc growth
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Epidermal growth factor receptor (EGFR) activation plays a pivotal role in EGFR-driven non-small cell lung cancer (NSCLC) and is considered as a key target of molecular targeted
therapy. EGFR tyrosine kinase inhibitors (TKIs) have been canonically used in NSCLC treatment. However, prevalent innate and acquired resistances and EGFR kinase-independent pro-survival
properties limit the clinical efficacy of EGFR TKIs. Therefore, the discovery of novel EGFR degraders is a promising approach towards improving therapeutic efficacy and overcoming drug
resistance. Here, we identified a 23-hydroxybetulinic acid derivative, namely DPBA, as a novel EGFR small-molecule ligand. It exerted potent in vitro and in vivo anticancer activity in both
EGFR wild type and mutant NSCLC by degrading EGFR. Mechanistic studies disclosed that DPBA binds to the EGFR extracellular domain at sites differing from those of EGF and EGFR. DPBA did not
induce EGFR dimerization, phosphorylation, and ubiquitination, but it significantly promoted EGFR degradation and repressed downstream survival pathways. Further analyses showed that DPBA
induced clathrin-independent EGFR endocytosis mediated by flotillin-dependent lipid rafts and unaffected by EGFR TKIs. Activation of the early and late endosome markers rab5 and rab7 but not
the recycling endosome marker rab11 was involved in DPBA-induced EGFR lysosomal degradation. The present study offers a new EGFR ligand for EGFR pharmacological degradation and proposes it
as a potential treatment for EGFR-positive NSCLC, particularly NSCLC with innate or acquired EGFR TKI resistance. DPBA can also serve as a chemical probe in the studies on EGFR trafficking
and degradation. SIMILAR CONTENT BEING VIEWED BY OTHERS JMJD5 INHIBITS LUNG CANCER PROGRESSION BY FACILITATING EGFR PROTEASOMAL DEGRADATION Article Open access 09 October 2023 TRIB3-EGFR
INTERACTION PROMOTES LUNG CANCER PROGRESSION AND DEFINES A THERAPEUTIC TARGET Article Open access 21 July 2020 FBXL2 COUNTERACTS GRP94 TO DESTABILIZE EGFR AND INHIBIT EGFR-DRIVEN NSCLC
GROWTH Article Open access 11 October 2021 INTRODUCTION Lung cancer is the most common cancer and the leading cause of cancer-related death, and non-small cell lung cancer (NSCLC) accounts
for 84% of all lung cancer diagnoses.1 Epidermal growth factor receptor (EGFR) is often overexpressed in NSCLC.2 EGFR tyrosine kinase inhibitors (TKIs) have been widely used to treat
EGFR-positive NSCLC. Although EGFR TKIs have demonstrated remarkable clinical efficacy, they may also invariably induce acquired resistance. Most patients present with a T790M mutation after
gefitinib treatment.3 The second-generation EGFR TKI afatinib irreversibly binds to EGFR, but it lacks selectivity for EGFR WT and EGFR T790M and provokes adverse reactions.4 The
third-generation EGFR TKI osimertinib is effective in patients with the T790M mutation.5 However, certain patients develop other acquired resistances such as a C797S mutation.6 The
inhibition of EGFR kinase activity may result in “kinome rewiring”, which, in turn, causes compensatory feedback activation of alternative kinases.7 Moreover, 10–20% of NSCLCs with EGFR
mutation are insensitive to EGFR TKIs and most NSCLCs with EGFR WT do not respond to TKIs despite EGFR up-regulation. This response is a manifestation of innate resistance.8,9 Recent studies
reveal that EGFR kinase-independent activity also promotes cancer cell survival and chemoresistance.10,11,12 Hence, targeting EGFR by inducing degradation rather than inhibition of kinase
activity could be a more effective and complete approach to repress EGFR in NSCLC treatment. Several EGFR-degrading strategies have been reported. One approach is to deliver specific EGFR
small interfering RNAs (siRNAs) in vivo. However, their short half-life, rapid degradability, and off-target effects have reduced their relative efficacy.13,14 A more promising strategy is
to target EGFR degradation with chemicals. Proteolysis-targeting chimera (PROTAC) technology has been used in EGFR degradation and has entailed the connexion of EGFR TKIs with E3 ligase
ligands to form ternary chimeras for proteasomal degradation.15,16 However, the optimization of solubility, membrane permeability, and metabolic stability increase challenge to make these
ternary chimeras druggable.17 EGFR degradation has been induced by several small molecules, although they do not directly target EGFR. Sanguinarine up-regulated NOX3 to elevate the reactive
oxygen species (ROS) level, resulting in EGFR oxidation and degradation.18 Curcumin and the surviving inhibitor YM-155 degraded EGFR by inducing the ubiquitin-proteasomal pathway.19,20
Autophagic degradation of EGFR was involved in cancer cell death caused by arsenic and celastrol.21,22 Inhibiting canonical EGFR endocytosis by the clathrin inhibitor pitstop2 rerouted EGFR
degradation to a macropinocytosis-mediated lysosomal pathway.23 The foregoing reports indicate that the mechanisms of EGFR endocytosis and degradation mediated by small molecules are complex
and stimulus-dependent. Thus, the discovery of novel EGFR degraders and the exploration of EGFR degradation mechanisms are critical in the development of new strategies to control
EGFR-positive NSCLC. Up to now, few small-molecule ligands directly targeting EGFR and inducing EGFR degradation have been demonstrated. Here, we identify a new small-molecule EGFR ligand
called DPBA, which functions as an EGFR degrader to inhibit the survival of EGFR-positive NSCLC. Specifically, DPBA binds to the EGFR extracellular domain (ECD) and induces
flotillin-1-mediated EGFR endocytosis and lysosomal degradation without EGFR dimerization, phosphorylation, or ubiquitination. DPBA has the potential to be developed into a new drug for
EGFR-positive NSCLC. RESULTS DPBA REDUCES THE VIABILITY OF NSCLC CELLS BY SUPPRESSING EGFR PROTEIN EXPRESSION AND THE DOWNSTREAM PATHWAYS EGFR degraders were screened from >700 natural
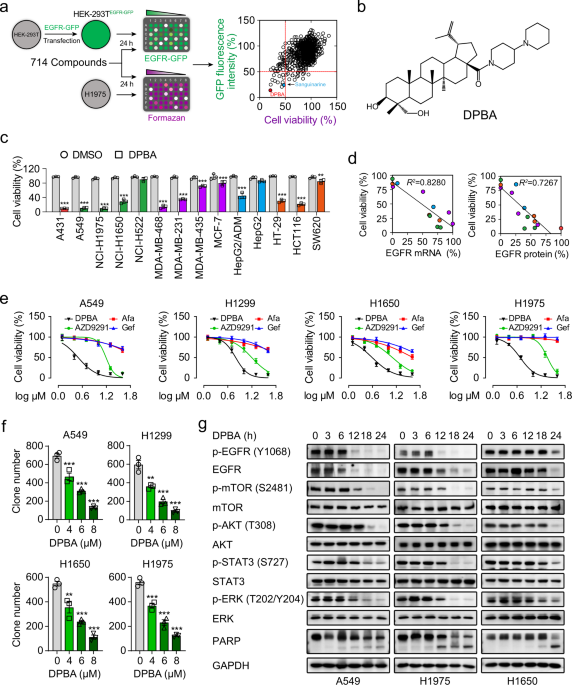
compounds and their derivatives (Supplementary Table 1). The screening process is described in the form of a flowchart in Fig. 1a. After test compounds’ treatment, green fluorescent protein
(GFP) fluorescence intensity of HEK-239TEGFR-GFP cells and viability of H1975 cells are determined, respectively. The well-defined EGFR degrader sanguinarine18 was the positive control (Fig.
1a). The most potent substance was compound 187, which is a derivative of 23-hydroxybetulinic acid (23-HBA), namely DPBA (Fig. 1b), synthesized by our group previously.24 DPBA
down-regulated only EGFR protein level among ErbB family members (Supplementary Fig. 1a), indicating that DPBA is an EGFR-specific inhibitor. We assessed the anticancer activity of DPBA (5
μM) on a panel of cancer cell lines in order to validate the association between the anticancer activity of DPBA and the EGFR levels. A431, A549, H1650, H1975, MDA-MB-468, and HCT116 with
high EGFR expression levels were the most sensitive to DPBA (Fig. 1c). Cancer cell sensitivity to DPBA was positively correlated with the EGFR messenger RNA (mRNA) and protein levels (Fig.
1d). We then determined the anticancer efficacy of DPBA against EGFR-positive NSCLC cell lines. The survival of A549 (EGFR WT), H1299 (EGFR WT), H1650 (del E736-A750), and H1975
(L858R/T790M) were suppressed to a greater extent by DPBA than by EGFR TKIs (gefitinib, afatinib, and AZD9291) (Fig. 1e). DPBA presented with lower cytotoxicity to human bronchial epithelial
cells (BEAS-2B) and immortalized human keratinocytes (HaCaT) than NSCLC cell lines (Supplementary Fig. 1b). The colony formation assay confirmed the antiproliferation effect of DPBA on
NSCLC cells (Fig. 1f). As the EGFR-driven pro-survival pathway is vital to tumour progression, we evaluated the effect of DPBA on EGFR downstream signalling pathway. DPBA significantly
down-regulated EGFR protein in EGFR WT and EGFR mutant NSCLC cell lines in a time-dependent manner and did not increase EGFR phosphorylation (Fig. 1g). DPBA also reduced downstream p-mTOR,
p-Akt, p-STAT3, and p-ERK phosphorylation and promoted the cleavage of the apoptotic marker PARP (poly (ADP-ribose) polymerase) (Fig. 1g). BEAS-AB and HaCaT cells treated with DPBA at the
same concentrations presented with no EGFR protein inhibition (Supplementary Fig. 1c). Therefore, EGFR protein down-regulation blocked the EGFR-driven pro-survival pathway and conferred DPBA
with anti-NSCLC activity. DPBA DECREASES EGFR PROTEIN LEVELS BY LYSOSOMAL DEGRADATION Protein expression may be reduced by the inhibition of de novo synthesis or by accelerated degradation.
DPBA did not down-regulate EGFR mRNA (Fig. 2a). The cycloheximide (CHX) chase assay showed that the DPBA plus CHX treatment lowered the EGFR protein levels considerably more than the CHX
treatment alone (Fig. 2b), indicating that DPBA down-regulates EGFR via protein degradation. Proteasomal and lysosomal degradation are two key protein degradation pathways. Here, DPBA did
not induce EGFR ubiquitination (Fig. 2c). The lysosome inhibitor bafilomycin A1 (baf A1) but not the proteasome inhibitor MG132 reversed DPBA-induced EGFR degradation (Fig. 2d). This
mechanism was confirmed with the lysosomal protease inhibitors leupeptin, E-64, Ca074Me, and pepstatin A (Fig. 2e). Baf A1 also reversed DPBA-induced inhibition of EGFR downstream signalling
and cell death in A549 and H1975 (Fig. 2f, g). These data suggest that a lysosomal degradation pathway mediates DPBA-induced EGFR protein decrease. RAB5 AND RAB7 ACTIVATION IS INVOLVED IN
DPBA-INDUCED ENDO-LYSOSOMAL TRAFFICKING OF EGFR The induction of endocytosis is a key step in EGFR degradation. We observed DPBA-induced perinuclear EGFR cluster formation (Fig. 2h). Next,
we conducted a surface biotinylation assay to establish that the perinuclear clusters were derived from intracellular or membrane-bound EGFR. As shown in Fig. 2i, surface EGFR was
significantly decreased, whereas plasma membrane-bound EGFR labelled with NHS-SS-biotin had substantially increased in the cytoplasm after DPBA treatment, indicating that DPBA induces
surface EGFR endocytosis. Endocytic EGFR may be transported to lysosomes via early and late endosomes, enter recycling endosomes and returned to the plasma membranes, or delivered to the
Golgi apparatus, endoplasmic reticulum, mitochondria or nuclei, depending on the stimulating factors.25 DPBA did not induce EGFR colocalisation with autophagosomes (LC-3-positive),
mitochondria (TOM20-positive), Golgi apparatus (GM130-positive), or endoplasmic reticulum (calnexin-positive) (Supplementary Fig. 2a). However, DPBA-induced endocytic EGFR colocalised with
Rab5 after 3 h and entered the late endosomes (Rab7-positive) and lysosomes (LAMP1-positive) after 6 h. No endocytic EGFR colocalisation with recycling endosomes (Rab11-positive) was
detected (Fig. 2j). Consistently, DPBA increased the Rab5-GTP and Rab7-GTP levels, but not the Rab11-GTP level (Fig. 2k). Collectively, these data demonstrate that DPBA-induced endocytic
EGFR get the label of lysosome degradation rather than being recycled to the plasma membranes. DPBA INDUCES EGFR ENDOCYTOSIS VIA A CLATHRIN-INDEPENDENT LIPID RAFT EGF-induced EGFR
endocytosis is primarily mediated by clathrin and dependent on tyrosine kinase activity.26,27 As expected, the suppression of EGFR activation with gefitinib or cetuximab or the inhibition of
clathrin with pitstop2 or clathrin siRNA blocked EGF-induced EGFR endocytosis (Fig. 3a, Supplementary Fig. 2b). However, gefitinib, AZD9291, pitstop2, or clathrin siRNA did not suppress
DPBA-induced EGFR endocytosis (Fig. 3b). DPBA treatment also degraded kinase-dead EGFR (Fig. 3c). These data clearly show that DPBA-induced EGFR endocytosis is clathrin independent and does
not require EGFR tyrosine kinase activity. However, EGFR still has serine/threonine phosphorylation sites, which also play important roles in EGFR regulation, including EGFR
endocytosis.28,29 We found that DPBA had no effect on EGFR serine/threonine phosphorylation level, indicating that Ser/Thr phosphorylation may not be involved in DPBA-induced EGFR
endocytosis (Supplementary Fig. 2c). Clathrin-independent EGFR endocytosis is achieved by a lipid raft-dependent process or macropinocytosis.23,30 The macropinocytosis inhibitor amiloride
blocked fluorescein isothiocyanate-dextran (FITC-dextran) uptake, but failed to block DPBA-induced EGFR endocytosis (Supplementary Fig. 2d). Here, the cholesterol extractor
methyl-β-cyclodextrin (MCD) substantially blocked the EGFR endocytosis (Fig. 3b), downstream pathway down-regulation, and cell death induced by DPBA (Fig. 3d, e). As cholesterol is an
important lipid raft constituent, the foregoing results indicate that DPBA-induced EGFR endocytosis is mediated by lipid rafts. It was further confirmed by the fact that DPBA induced a
significant EGFR residence in the lipid raft domain (Fig. 3f). Taken together, our results demonstrate that the lipid raft microdomain serves as an organizational platform for the initiation
of DPBA-induced EGFR endocytosis. DPBA-INDUCED EGFR ENDOCYTOSIS DEPENDS ON FLOTILLIN-1 Lipid raft-dependent endocytosis is mediated either by caveolin or flotillin or by small guanosine
triphosphatases, such as GRAF1, Arf6, or RhoA.31 Here, only flotillin-1 knockdown blocked DPBA-induced EGFR endocytosis and degradation (Fig. 3g, h, Supplementary Fig. 2e). In
flotillin-1-mediated endocytosis, flotillin-1 is recruited to the surface cargo and forms pre-endocytic clusters.32 DPBA treatment also enhanced the interaction between flotillin-1 and EGFR
(Fig. 3i). As dynamin 2 catalyses endocytic vesicle scission,31 we investigated the role of dynamin 2 in DPBA-induced EGFR endocytosis and degradation. Dynamin 2 siRNA did not recover
DPBA-induced EGFR endocytosis or degradation (Fig. 3g, j). This mechanism was confirmed with the potent dynamin 2 inhibitor dyngo-4a (Supplementary Fig. 2f). Thus, flotillin-1 is a key
moderator in DPBA-induced EGFR endocytosis and the latter is independent of dynamin 2. DPBA DIRECTLY BINDS TO EGFR ECD AND TRIGGERS EGFR DEGRADATION As the ligand EGF binds to EGFR and
induces EGFR activation and degradation, we attempted to determine whether DPBA has a similar mode of action. We used cellular thermal shift assay to test the cell-level interaction between
DPBA and EGFR. Relative to the control, EGFR showed thermal shifts in the presence of DPBA at the denaturation temperature range of 37–58 °C (Fig. 4a), indicating that DPBA may directly bind
to EGFR and thermally stabilize it in vivo. We then performed BIAcore kinetics and microscale thermophoresis (MST) analyses on binding between recombinant EGFR ECD and DPBA, and EGF was the
positive control. The BIAcore assay showed that DPBA bound to EGFR ECD, and the association constant (_K_d) was 39.5 μM (Fig. 4b, Supplementary Fig. 3a). However, the curve disclosed
distinct binding kinetics between DPBA and EGF, indicating that each has its unique binding characteristic with EGFR. The MST assay revealed that DPBA interacted with EGFR ECD in a
dose-dependent manner and _K_d = 38.4 ± 1.75 μM, whereas _K_d = 1.92 ± 0.18 μM for EGF. However, the presence of DPBA did not affect the interaction between EGFR ECD and EGF as the _K_d did
not markedly change (3.14 ± 0.42 μM) (Fig. 4c). These data imply that DPBA and EGF have different binding sites for EGFR. DPBA did not interact with the intracellular domain (ICD) of EGFR,
or the ECD of Her2, Her3, or Her4 (Supplementary Fig. 3b). Therefore, DPBA specifically binds to EGFR ECD. The parent compound of DPBA (23-HBA) did not bind to EGFR ECD and had no effect on
the EGFR protein level (Supplementary Fig. 3b, c), suggesting that the dipiperidine group may be an absolute requirement for the interaction between DPBA and EGFR. To further confirm the
interaction between DPBA and EGFR, we synthesized the probe DPBA-1, using a photoaffinity-labelling technique by introducing a tetrazole-containing base with an alkynyl terminal to the
inactive C-23 site. A negative probe (NP) was used as the negative control (Supplementary Fig. 3d). The introduction of the tetrazole-containing base had no influence on cytotoxicity and
EGFR degradation of DPBA (Supplementary Fig. 3e, f). The pull-down assay showed that DPBA-1 interacted with EGFR in a dose-dependent manner in the intact cells and cell lysates. Excess DPBA
(10×) significantly inhibited the interaction between DPBA-1 and EGFR, indicating that both competed for the same site (Fig. 4d). Neither EGF nor cetuximab interfered with the interaction
between EGFR and DPBA-1 (Fig. 4e). This observation suggests that DPBA binds to sites different from those of EGF and cetuximab. EGF binding to EGFR induced EGFR dimerization and activation,
whereas DPBA did neither (Fig. 4f, g). Thus, DPBA and EGF differ in terms of binding sites and mechanisms. We next checked whether EGFR ECD binding was crucial for DPBA-induced EGFR
degradation. As shown in Fig. 4h, EGFR ECD deletion (del 25–645) blocked DPBA-induced EGFR degradation. Collectively, these results indicate that DPBA specifically binds to EGFR ECD to
trigger EGFR degradation. DPBA INHIBITS EGFR WT AND EGFR MUTANT NSCLC GROWTH IN VIVO To examine the anti-NSCLC activity of DPBA in vivo, we established the tumour xenografts models of A549,
H1650, and H1975 cells, and a xenograft derived from a patient with primary EGFR-positive lung cancer (Supplementary Fig. 4a). Mice treated with DPBA (25 mg/kg) presented with considerable
tumour growth inhibition, but there was no change in body weight (Fig. 5a–d, Supplementary Fig. 4b, c). Haematoxylin–eosin (H&E) staining revealed that DPBA induced substantial cell
death in the tumour sections. Ki67, a marker of proliferation and EGFR expression levels considerably decreased after DPBA treatment (Fig. 5e, f). The EGFR pathway was markedly repressed in
the DPBA-treated patient-derived xenograft (PDX) model (Fig. 5g). To assess DPBA toxicity, we collected mouse blood and organs for serum biochemistry, routine blood analyses, and
pathological examination, respectively. As shown in Supplementary Fig. 4d, the white blood cells, red blood cells, platelets, and haemoglobin counts did not significantly change in response
to DPBA exposure. Lactate dehydrogenase, creatine kinase, alanine transaminase, aspartate aminotransferase, creatinine, blood urea nitrogen, and spleen weight were not significantly altered
by DPBA treatment. H&E staining of the kidney, spleen, liver, and heart revealed that DPBA had no significant toxic effects on any of them (Supplementary Fig. 4e). Overall, DPBA induced
EGFR degradation and markedly inhibited the growth of EGFR-positive NSCLC xenografts with negligible toxicity. DISCUSSION Due to the inevitable acquired resistance and kinase-independent
functions of EGFR, targeting EGFR degradation by small molecules is a promising cancer treatment strategy.33 Here, we identified the 23-HBA derivative DPBA that induces EGFR degradation by
directly binding to EGFR ECD, blocking downstream pathways and thereby suppressing NSCLC growth. Mechanistically, DPBA binding to EGFR ECD differs from that between EGFR and TKIs. Thus, DPBA
induces EGFR degradation and cell death regardless of the mutations in the ICD, such as del E746-A750, L858R, and T790M. Unlike EGF or a reported small-molecule EGFR ligand NSC228155,34
DPBA neither induces EGFR dimerization nor activates EGFR and its downstream pathways. In this way, it avoids the pro-survival effects of EGFR activation. Moreover, DPBA does not alter EGFR
expression in normal human cells. DPBA potently inhibits the growth of several EGFR-positive NSCLC xenografts and has negligible toxicity. Overall, DPBA is a novel EGFR degrader that could
potentially treat both EGFR WT and EGFR mutant NSCLC. EGFR ICD initiates the EGFR signalling pathway and is a major EGFR target. EGFR TKIs and PROTAC EGFR degraders target this region. EGFR
degradation was also achieved by targeting the allosteric site of the EGFR kinase domain by weak reversible EGFR inhibitors without connecting of proteasomal degradation tags to EGFR TKIs.35
The aforementioned strategies are effective and specific to achieve EGFR degradation. However, most EGFR mutations focus on the kinase domain.36 Thus, inducing EGFR degradation by targeting
the kinase region may not avoid EGFR mutations. An alternative approach to EGFR degradation is to target the ECD. It has been reported recently that T315 initiates EGFR degradation by
binding the ECD. Its action depends on Y1045 phosphorylation and CBL recruitment. CBL is an E3 ubiquitin-protein ligase that drives EGFR proteasomal degradation.37 This process aligns with
the conventional EGF-induced EGFR degradation mechanism and indicates that T315 may function in a way similar to that of EGF.38 DPBA did not induce EGFR phosphorylation, dimerization, or
ubiquitination, and EGFR TKIs and MG132 did not rescue EGFR degradation. Thus, DPBA regulates EGFR in a heretofore unknown manner. Furthermore, we performed de novo liquid chromatography
mass spectrometry (LC-MS) sequencing analysis using a photoaffinity-labelled probe DPBA-1, and identified three potential sites, D166, D303, and D321. The alkaline dipiperidine group in DPBA
may be important for the binding to the acidic amino acids of EGFR. Site-directed mutagenesis experiments need be further conducted on EGFR to localize its key binding sites. Besides, the
crystalline structure of EGFR bound to DPBA will be helpful to reveal the binding sites and conformation of EGFR with DPBA. Given that DPBA-induced EGFR degradation is resistant to EGFR
TKIs, EGFR kinase-independent functions may be involved. These non-canonical EGFR functions are mostly achieved by interacting with other chaperone proteins, such as HSP90, CDC34, and SGLT1.
These interaction levels are higher in tumour cells than in normal cells.10,39,40,41,42,43,44 DPBA may destabilize EGFR through interrupting these interactions. This may partially explain
why DPBA showed specific toxicity towards tumour cells. Interaction proteomics is required to analyse potential interaction partners and reveal the degradation mechanism of EGFR in tumour
cells upon DPBA binding. It has been reported that EGFR dimerization not only induces autophosphorylation but also maintains EGFR stability. Impairing EGFR dimerization by a specific peptide
can induce EGFR degradation.45 Whether DPBA has a similar potency needs further investigation. Nevertheless, the discovery of DPBA as a novel EGFR ligand underscores the fact that EGFR may
be degraded by targeting EGFR ECD. Elucidation of the binding and interaction mechanisms between DPBA and EGFR will help guide the design of new EGFR degraders that selectively target EGFR
ECD. The endocytic mechanism and post-endocytic fates of EGFR are complex and stimulus-dependent.25,26 Canonical EGF-induced EGFR endocytosis is regulated by both clathrin-mediated
endocytosis (CME) and non-CME (NCE). CME is activated at all EGF concentrations, whereas NCE is induced only at high EGF concentrations wherein the receptor is internalized via lipid
rafts.46 Ligand-independent EGFR endocytosis has also been detected in response to various stimuli such as chemotherapeutic agents, ultraviolet, EGFR TKIs, EGFR monoclonal antibodies and
ROS.25 EGFR TKIs induced the internalization of inactivated EGFR and endosomal arrest, which is considered an innate TKI resistance mechanism.47 Ultraviolet (UV) or cisplatin-induced
clathrin-mediated EGFR internalization, which entails serine and threonine phosphorylation catalysed by p38 kinase.48,49 Cetuximab stimulated caveolin-mediated EGFR endocytosis that was
independent of tyrosine kinase activity but mediated by p38.50,51,52 CHX-induced EGFR endocytosis was also p38-dependent, but tyrosine kinase-independent.53 It seems that EGFR tyrosine
kinase activity is expendable, whereas p38 plays a key role in EGFR endocytosis. DPBA-induced EGFR endocytosis is also tyrosine kinase independent, but the p38 inhibitor SB203580 did not
block EGFR endocytosis (data not shown). This finding is consistent with that of a previous study, which showed that p38-mediated endocytic EGFR is recycled back to the plasma membrane.54
Here, we revealed a novel DPBA-initiated EGFR endocytosis mechanism that is mediated by a flotillin-dependent lipid raft and does not involve EGFR dimerization, phosphorylation, and
ubiquitination. This unique mechanism enables DPBA to serve as a new chemical probe in the ongoing exploration of EGFR endocytosis and trafficking. 23-HBA, a lupane-type pentacyclic
triterpene extracted from the Chinese medicinal herb _Pulsatilla chinensis_ by our group, has been found to exhibit anti-tumour activity in vitro and in vivo. In order to improve its
biological activity, we carried out structural modifications and obtained a series of derivatives with improved activities.24 The anti-tumour mechanisms of 23-HBA or its derivatives mainly
involve mitochondrial ROS burst, mitochondrial membrane potential depolarization, non-classical mitochondrial autophagy, telomerase activity inhibition, and multidrug resistance
reversal.55,56,57,58 So far, there have been no reports on 23-HBA or its derivatives, whose modes of action resembles that of DPBA. Our study sheds a new insight into the molecular mechanism
of 23-HBA and its derivatives. The present study identifies one 23-HBA derivative, DPBA, a promising anticancer drug candidate that binds to EGFR ECD and promotes EGFR endocytosis and
lysosomal degradation in the treatment of EGFR-positive NSCLC (Fig. 5h). Here, we unveiled a new class of small-molecule EGFR degraders directly targeting EGFR ECD. In this way, we provide a
strategy to inhibit EGFR kinase-independent functions and suppress innate or acquried EGFR TKI-mediated NSCLC resistance. MATERIALS AND METHODS CELL CULTURE A431, A549, NCI-H1299,
NCI-H1650, NCI-H1975, NCI-H522, MDA-MB-231, MDA-MB-435, MDA-MB-453, MDA-MB-468, MCF-7, HepG2, HT-29, HCT116, SW620, HEK-293T, and BEAS-2B were purchased from the American Type Culture
Collection (Manassas, VA, USA). HaCaT was obtained from Beina Chuanglian Biotechnology Research Institute (Beijing, China). HepG2/ADM cells were generously provided by Prof. Kwok-Pui Fung
(Chinese University of Hong Kong, Hong Kong, China). H1299, H1650, and H1975 were cultured in RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) foetal
bovine serum (FBS, Thermo Fisher Scientific) and 1% (v/v) penicillin–streptomycin (PS, Thermo Fisher Scientific). All other cell lines were cultured in Dulbecco’s modified Eagle’s medium
supplemented with 10% (v/v) FBS and 1% (v/v) PS. Cells were maintained at 37 °C in a humidified atmosphere incubator with 5% CO2. Cell line authentication and detection of mycoplasma
contamination were performed before usage. REAGENTS AND ANTIBODIES DPBA (98% purity) was synthesized as described previously.24 DAPI (4′,6-diamidino-2-phenylindole), CHX, leupeptin, E-64,
Ca074Me, pepstatin A, pitstop2, MCD, FITC-dextran, Tris (2-carboxyethyl) phosphine hydrochloride (TCEP), Tris [(1-benzyl-1_H_-1,2,3-triazol-4-yl) methyl] amine (TBTA), Biotin-N3 and
Azide-fluor 545 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Hoechst 33342, BS3 (bis (sulfosuccinimidyl) suberate), EZ-Link Sulfo-NHS-SS-Biotin, biotinamidohexanoic acid
_N_-hydroxysuccinimide ester, Pierce™ Avidin Agarose, a BCA Protein Assay Kit, and a DAB Kit were obtained from Thermo Fisher Scientific. Gefitinib, afatinib, AZD9291, MG132, Baf A1,
Amiloride HCl, and dyngo-4a were purchased from Selleck (Houston, TX, USA). Laemmli sample buffer (2×) and an ECL Chemiluminescence Detection Kit were purchased from Bio-Rad (Hercules, CA,
USA). Matrigel was obtained from BD Biosciences (San Jose, CA, USA). A Total RNA Kit was purchased from OMEGA BIO-TEK (Norcross, GA, USA). SYBR Green I Master and a Transcriptor First Strand
cDNA Synthesis Kit was obtained from Roche (Mannheim, Germany). Antibodies against PARP, cleaved PARP, EGFR (rabbit monoclonal), Her2, Her3, Her4, p-EGFR (Y1068), p-EGFR (Y1045), mTOR,
p-mTOR (S2481), STAT3, p-STAT3 (S727), Akt, p-Akt (T308), ERK, p-ERK (T202/204), phospho-threonine, transferrin receptor 1 (TfR), Rab5, Rab7, Rab11, LC-3, flotillin-1, caveolin-1, GRAF1,
Arf6, Ki67, β-actin, GAPDH, anti-rabbit IgG, and anti-mouse IgG were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against EGFR (mouse monoclonal), dynamin 2 and
RhoA were obtained from Abcam (Cambridge, MA, USA). Phospho-serine antibody was purchased from Merck Millipore (Darmstadt, Germany). All other reagents were purchased from Sigma-Aldrich.
PLASMID CONSTRUCTION AND TRANSFECTION EGFR-GFP was obtained from Addgene (#32751). pCMV-HA EGFR was constructed by cloning EGFR WT complementary DNA (cDNA) into the pCMV-HA vector. pCMV-HA
EGFR KD (K745A, kinase dead) was generated from pCMV-HA EGFR using KOD-Plus Mutagenesis Kit (Toyobo, Japan). Primers for EGFR KD were as follows: ATCGCGGAATTAAGAGAAGCAACAT (forward);
AGCGACGGGAATTTTAACTTTCTCA (reverse). EGFR ECD deletion (del 25–645) cDNA was synthesized by Beijing Genomics Institute (Beijing, China) and cloned into the pCMV-HA vector to generate pCMV-HA
EGFR Δ. All these plasmids were constructed by using the restriction sites _Sal_l and _Not_l. Transfections were performed by using Lipofectamine 3000 reagent according to the
manufacturer’s protocol. CELL VIABILITY ASSAY Cells with indicated treatment were incubated with MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2_H_-tetrazolium bromide; 5 mg/ml) for 4 h.
Dimethyl sulfoxide (DMSO) were added to solubilize the formazan crystals, and the absorbance was measured at 595 nm using a microplate reader (Beckman Coulter, Brea, CA, USA). Cell viability
was calculated as a percentage of the vehicle control group treated with a medium containing 0.2% DMSO. WESTERN BLOT Cells were lysed with RIPA lysis buffer containing protease inhibitor
cocktail. Thirty micrograms of total protein was separated on sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and then transferred onto polyvinylidene fluoride
membranes. The levels of indicated proteins were blotted by incubating with primary antibodies overnight at 4 °C, followed by incubation with secondary antibodies for 1 h at room temperature
(RT). Immunoreactive proteins were visualized using an ECL Chemiluminescence Detection Kit. COLONY FORMATION ASSAY Cells treated with indicated concentrations of DPBA for 24 h were
trypsinized and seeded in 6-well plates as a density of 0.8 × 103 cells/well and cultured for 10 days. At the end, cells were fixed with 4% paraformaldehyde (PFA) and stained with a 0.1%
crystal violet solution. Cell colonies were photographed by a CKX41 inverted microscope (Olympus, Japan) and counted using ImagePro Plus v. 6.0 (Media Cybernetics Inc., Rockville, MD, USA).
REVERSE TRANSCRIPTION POLYMERASE CHAIN REACTION (RT-PCR) ASSAY Two micrograms of total RNA were transformed to cDNA by a Transcriptor First Strand cDNA Synthesis Kit. RT-PCR was performed by
mixing 10 μl SYBR Green I Master, 0.5 μM forward primer, 0.5 μM reverse primer, 2 μl cDNA, and 6 μl distilled water per sample. The PCR products were quantified with a LightCycler 480 PCR
system (Roche, Mannheim, Germany). The primers used for PCR were synthesized by Beijing Genomics Institute and the sequences for EGFR were: 5′-CCTGGTCTGGAAGTACGCAG-3′ and
5′-CGATGGACGGGATCTTAGGC-3′. IMMUNOFLUORESCENCE ASSAY Cells with indicated treatments were fixed in 4% PFA and blocked in 5% bovine serum albumin containing 0.4% Triton X-100. Then, the cells
were incubated using the indicated primary antibody at 4 °C overnight, followed by a fluorescent secondary antibody for 1 h at RT and stained with 5 μg/ml of DAPI for 5 min. The cellular
fluorescence was photographed by a Zeiss AX10 microscope (Carl Zeiss, Göttingen, Germany). EGFR ENDOCYTOSIS ASSAY Surface and endocytic EGFR were localized by biotinylation assay.59 For the
surface EGFR, the cells were biotinylated with 0.5 mg/ml NHS-SS-biotin for 30 min at 4 °C, washed with ice-cold phosphate-buffered saline (PBS), and blocked with 50 mM glycine for 20 min.
For the endocytic EGFR, the cells were biotinylated with NHS-SS-biotin and blocked with glycine before the DPBA treatment. Surface NHS-SS-biotin EGFR was reduced with 50 mM glutathione in 90
mM NaCl, 1 mM MgCl2, 0.1 mM CaCl2, 60 mM NaOH, and 10% (v/v) FBS for 30 min. The cells were then lysed and the biotinylated EGFR was precipitated with avidin agarose beads for 2 h at 4 °C.
The EGFR was detected by Western blot. RAB-GTP ACTIVITY ANALYSIS Activation of Rab5, Rab7, and Rab11 were analysed with Rab5, Rab7, and Rab11 Activation Assay Kits (NewEast Biosciences, King
of Prussia, PA, USA). Briefly, 500 μg total protein lysed with 1× lysis buffer per sample were used in a Rab-GTP pull-down assay. Each sample was combined with anti-Rab-GTP monoclonal
antibody and incubated at 4 °C overnight. Protein A/G agarose beads were used to capture the antibodies. After 1 h incubation at 4 °C, the beads were boiled in 2× loading buffer for 5 min.
The supernatant was used for Western blot to detect Rab-GTP activity using Rab polyclonal antibody. SIRNA TRANSFECTION ASSAY Cells were transfected with control siRNA duplexes or specific
siRNA duplexes with indicated targets using Lipofectamine 3000. After transfection for 48 h, cells were exposed to indicated treatment, and the expression levels of specific proteins were
measured by Western blot. siRNAs were synthesized by GenePharma (Shanghai, China) and the sequences were shown in Supplementary Table 2. PREPARATION OF DETERGENT-FREE LIPID RAFTS
Detergent-free lipid rafts were prepared as described previously.60 After washing with ice-cold PBS, cells were resuspended in 0.5 ml lysis buffer (20 mM Tis-HCl, 250 mM sucrose, 1 mM CaCl2,
1 mM MgCl2, pH = 7.8) with protease inhibitor cocktail and lysed by passing through a 22-G needle 40 times. The post-nuclear supernatant was collected by centrifugation at 1000 × _g_ for 10
min, while the precipitated pellet was lysed again in the same way in 0.5 ml lysis buffer and the post-nuclear supernatant was combined with the first. One millilitre of lysis buffer with
50% (v/v) OptiPrep was added to the supernatants and transferred to an ultra-centrifuge tube. Three millilitres of 0–20% gradient OptiPrep in lysis buffer was placed on the top of the
mixture. After centrifugation at 25,000 r.p.m. for 90 min in a Beckman ultra-centrifuge, 10 fractions were collected from top to bottom of the gradients. EGFR, TfR, flotillin-1, and
caveolin-1 in different fractions were analysed by Western blot. CETUXIMAB BIOTINYLATION Cetuximab biotinylation was performed as described previously.61 In brief, glycine was removed from
Erbitux by ultrafiltration in 0.9% NaCl. Cetuximab and biotinamidohexanoic acid _N_-hydroxysuccinimide ester dissolved in DMSO (1 mg/ml) were mixed in the ratio of 1:2. Two micrograms of
cetuximab without glycine was incubated with 12 μg biotinamidohexanoic acid _N_-hydroxysuccinimide ester at 4 °C overnight. Ten microlitres of 10× PBS was added to adjust pH to 7.0. The
process was stopped by adding 20 μl 2 M Tris and incubated at RT for 60 min. Excess biotinamidohexanoic acid _N_-hydroxysuccinimide was removed by ultrafiltration. CO-IMMUNOPRECIPITATION
Cells were lysed in immunoprecipitation buffer (Thermo Fisher) with protease inhibitor cocktail. EGFR was immunoprecipitated by incubating with biotinylated cetuximab at 4 °C overnight,
followed by incubation with avidin agarose beads at RT for 2 h. Immune complexes were washed five times with immunoprecipitation buffer, then eluted by boiling in 2× loading buffer for 5
min. The expression levels of interacting proteins were detected by Western blot. CELLULAR THERMAL SHIFT ASSAY Cells treated with DPBA were disrupted by liquid nitrogen, then centrifuged at
12,000 r.p.m. for 15 min to get total protein extracts. The proteins were divided into eight fractions and denatured at different temperatures for 10 min. Denatured proteins were
precipitated by centrifugation at 12,000 r.p.m. for 10 min. Supernatant undenatured EGFR was detected by Western blot. BIACORE Recombinant human EGFR ECD (Sino Biological, Beijing, China)
was directly coupled to CM-5 chip’s (GE Healthcare Life Sciences, Marlborough, MA, USA) different channel according to the isoelectric point of EGFR protein. The interaction between EGFR and
DPBA was measured by a BIAcore S200 (GE Healthcare Life Sciences). Affinity curve and kinetic curve were finally obtained using Biacore S200 Evaluation Software. EGF was used as a positive
control. MICROSCALE THERMOPHORESIS Recombinant human EGFR ECD, EGFR ICD, HER2 ECD, HER3 ECD, and HER4 ECD were obtained from Sino Biological Inc. (Beijing, China). They were labelled with a
Monolith NT Protein Labelling Kit RED-NHS (Nanotemper, Munich, Germany) and diluted to 250 nM with PBS. Label-free DPBA was diluted at half-concentrations with PBS (100,000–12.21 nM).
Label-free EGF was diluted at half-concentrations with PBS (8000–2 nM). Protein samples were mixed with DPBA or EGF and incubated at RT for 5 min. In a competitive assay, EGFR was incubated
with DPBA (100 μM) for 60 min at RT before EGF treatment (8000–2 nM). The mixtures were centrifuged at 16,000 × _g_ for 5 min and loaded into capillaries. Microscale thermophoresis
measurements were performed in a Monolith NT.115 (Nanotemper, Munich, Germany). SYNTHESIS OF DPBA-1 PROBE Synthesis of DPBA-1 was shown in Supplementary Fig. 3d. Briefly, HOBT (15 mg, 0.11
mmol), EDCI (22 mg, 0.11 mmol), and two drops of TEA were added to a solution of S1 (NP) (24.4 mg, 0.1 mmol). The mixture was stirred for 30 min at RT followed by the addition of DPBA (62.2
mg, 0.1 mmol). The reaction was then stirred at RT overnight in dark. Subsequently, the reaction was quenched by the addition of water and extracted with ethyl acetate. The organic layers
were combined, washed with brine and dried over anhydrous Na2SO4. Upon solvent evaporation in vacuo, the residue was purified by flash column (DCM:MeOH = 30:1) to give DPBA-1 as a white
solid (26 mg, 30% yield). 1H NMR (400 MHz, CDCl3) δ 8.22–8.01 (m, 2H), 7.21–7.04 (m, 2H), 4.78 (dd, _J_ = 5.2, 2.7 Hz, 2H), 4.70 (d, _J_ = 5.6 Hz, 1H), 4.59 (dd, _J_ = 15.2, 4.2 Hz, 2H),
4.24 (dd, _J_ = 47.7, 7.0 Hz, 1H), 3.63 (dd, _J_ = 9.6, 6.2 Hz, 1H), 3.31 (s, 1H), 3.17–2.51 (m, 9H), 2.29–2.09 (m, 2H), 1.97 (m, 5H), 1.74–1.61 (m, 5H), 1.54–1.18 (m, 14H), 0.99–0.79 (m,
10H). 13C NMR (101 MHz, CDCl3) δ 13C NMR (101 MHz, CDCl3) δ 173.70, 159.06, 158.95, 157.92, 151.12, 130.46, 127.59, 121.88, 115.91, 109.27, 73.50, 63.67, 57.27, 56.17, 55.12, 54.69, 53.10,
52.60, 51.59, 50.95, 49.27, 48.94, 47.40, 46.30, 45.60, 42.74, 41.87, 40.78, 38.36, 37.44, 36.89, 35.89, 34.82, 33.11, 32.47, 31.72, 30.38, 29.78, 28.30, 26.92, 25.89, 23.71, 22.91, 21.52,
20.21, 19.64, 18.71, 17.43, 16.83, 16.58, 16.13, 16.09, 14.75, 14.57, 13.73, 12.95 and 11.85. LC-MS (electrospray ionization) calculated for C51H73N6O5 [M + H] +: 849.5, found 849.5. EGFR
PULL-DOWN ASSAY The EGFR pull-down assay was performed with DPBA-1, an affinity-based probes.62 For total cell pull-down, the cells were treated with DPBA-1, subjected to UV irradiation (302
nm) for 10 min, and lysed with RIPA buffer containing a protease inhibitor cocktail. Then, 200 μg total protein (1 mg/ml) was used in a click chemistry reaction with biotin-N3 (50 μM), TBTA
(100 μM), TCEP (1 mM), and CuSO4 (1 mM), and incubated for 2 h at RT. For the cell lysate pull-down, the cells were lysed by alternate freeze-thaw in liquid nitrogen. Then, 200 μg total
protein (1 mg/ml) was incubated with DPBA-1 in the presence or absence of 10× DPBA for 60 min and subjected to UV irradiation (302 nm), and a click chemistry reaction. The reaction was
stopped by adding pre-chilled acetone (−20 °C) to precipitate the proteins. After centrifugation at 12,000 r.p.m. and 4 °C for 10 min, the precipitates were dissolved in PBS containing 1%
(v/v) SDS and incubated with streptavidin beads for 2 h at RT. The pull-down proteins were eluted with 2× loading buffer at 100 °C for 5 min. The EGFR level was detected by Western blot.
EGFR DIMERIZATION ASSAY EGFR dimerization was detected by cross-linking assay.63 Briefly, cells treated with EGF or DPBA at 4 °C (inhibition of EGFR endocytosis) were incubated with 0.5 mM
BS3 at 4 °C for 60 min. The reaction was quenched by incubating with 100 mM glycine at 4 °C for 15 min. Then, cells were lysed in RIPA lysis buffer with protease inhibitor cocktail and EGFR
monomer and dimer were separated by 5% SDS-PAGE and detected by Western blot. TUMOUR XENOGRAFTS IN NUDE MICE The in vivo experiments were approved by the Laboratory Animal Ethics Committee
of Jinan University (Guangzhou, China) (No. 201949-02). Primary surgical lung cancer specimens were obtained from the First Affiliated Hospital of Jinan University. Patient data are listed
in Supplementary Table 3. Fresh lung cancer tumours were cut into 3–5 mm3 sections and subcutaneously inoculated into the axillae of 8-week-old male NOD/SCID mice procured from GemPharmatech
Co. Ltd. (Jiangsu, China). The A549, H1650, and H1975 tumour xenograft models were established by suspending 107 cells from each cell line in Matrigel plus PBS at a 2:1 volumetric ratio and
subcutaneously injecting the suspensions into the axillae of 6-week-old BALB/c nude mice acquired from Vital River Laboratory Animal Technology (Beijing, China). The mice were randomly
divided into the vehicle and DPBA treatment groups when the tumour volumes reached ~100 mm3. Vehicle (3% (v/v) DMSO in deionised water) or 25 mg/kg DPBA dissolved in deionised water
containing 3% (v/v) DMSO was administered intragastrically once daily for ~1 month. Tumour volumes and body weights were measured every other day. Tumour volumes were calculated as (_a_ ×
_b_2)/2, where _a_ and _b_ are the longest and shortest tumour diameters, respectively. At the end of the experiments, the mice were anesthetised by intraperitoneal injection of 5 ml/kg of
1% (v/v) pentobarbital sodium salt. Their tumours were excised and their blood was drawn for the subsequent measurements. HISTOLOGY AND IMMUNOHISTOCHEMISTRY (IHC) For the H&E staining,
the tumour tissues were fixed in 4% (v/v) PFA, embedded in paraffin, sliced into 5-µm sections, and stained with H&E. For the IHC assay, the tumour sections were incubated with anti-Ki67
and anti-EGFR antibodies overnight at 4 °C, followed by horse radish peroxidase-conjugated secondary antibodies. The sections were visualized with a DAB Kit and the images were observed
under an Olympus BX 53 microscope (Olympus Corp., Tokyo, Japan). All images were statistically analysed in ImagePro Plus v. 6.0 (Media Cybernetics Inc., Rockville, MD, USA). STATISTICAL
ANALYSIS Each experiment was performed at least three times, and the data were shown as the mean ± standard deviation. Significant differences between two groups were determined using the
two-tailed unpaired _t_ test, and one-way analysis of variance, followed by Tukey’s post hoc test was used to evaluated significant differences between more than two groups. Differences were
considered significant when _P_ < 0.05. All statistical data were calculated using the GraphPad Prism 6.0 software. DATA AVAILABILITY The information of 714 compounds is enclosed in
Supplementary Table 1. The original datasets are also available from the corresponding author upon request. REFERENCES * Molina, J. R. et al. Non-small cell lung cancer: epidemiology, risk
factors, treatment, and survivorship. _Mayo Clin. Proc._ 83, 584–594 (2008). PubMed Google Scholar * Pao, W. & Chmielecki, J. Rational, biologically based treatment of EGFR-mutant
non-small-cell lung cancer. _Nat. Rev. Cancer_ 10, 760–774 (2010). CAS PubMed PubMed Central Google Scholar * Mok, T. S. et al. Gefitinib or carboplatin–paclitaxel in pulmonary
adenocarcinoma. _N. Engl. J. Med._ 361, 947–957 (2009). CAS PubMed Google Scholar * Yu, H. A. & Pao, W. Targeted therapies: Afatinib—new therapy option for EGFR-mutant lung cancer.
_Nat. Rev. Clin. Oncol._ 10, 551–552 (2013). CAS PubMed PubMed Central Google Scholar * Cross, D. A. et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR
inhibitors in lung cancer. _Cancer Discov._ 4, 1046–1061 (2014). CAS PubMed PubMed Central Google Scholar * Thress, K. S. et al. Acquired EGFR C797S mutation mediates resistance to
AZD9291 in non-small cell lung cancer harboring EGFR T790M. _Nat. Med._ 21, 560–562 (2015). CAS PubMed PubMed Central Google Scholar * Graves, L. M., Duncan, J. S., Whittle, M. C. &
Johnson, G. L. The dynamic nature of the kinome. _Biochem. J._ 450, 1–8 (2013). CAS PubMed Google Scholar * Gelsomino, F. et al. Epidermal growth factor receptor tyrosine kinase inhibitor
treatment in patients with EGFR wild-type non-small-cell lung cancer: the never-ending story. _J. Clin. Oncol._ 31, 3291–3293 (2013). PubMed Google Scholar * Itchins, M., Clarke, S. &
Pavlakis, N. Do EGFR tyrosine kinase inhibitors (TKIs) still have a role in EGFR wild-type pre-treated advanced non-small cell lung cancer (NSCLC)?—the shifting paradigm of therapeutics.
_Transl. Lung Cancer Res._ 7, S39–S45 (2018). CAS PubMed PubMed Central Google Scholar * Weihua, Z. et al. Survival of cancer cells is maintained by EGFR independent of its kinase
activity. _Cancer Cell_ 13, 385–393 (2008). PubMed PubMed Central Google Scholar * Che, T. F. et al. Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes
metastasis in NSCLC. _Oncotarget_ 6, 37349–37366 (2015). PubMed PubMed Central Google Scholar * Cao, X., Zhu, H., Ali-Osman, F. & Lo, H. W. EGFR and EGFRvIII undergo stress- and EGFR
kinase inhibitor-induced mitochondrial translocalization: a potential mechanism of EGFR-driven antagonism of apoptosis. _Mol. Cancer_ 10, 26 (2011). CAS PubMed PubMed Central Google
Scholar * Dykxhoorn, D. M., Palliser, D. & Lieberman, J. The silent treatment: siRNAs as small molecule drugs. _Gene Ther._ 13, 541–552 (2006). CAS PubMed Google Scholar * Tam, Y.
Y., Chen, S. & Cullis, P. R. Advances in lipid nanoparticles for siRNA delivery. _Pharmaceutics_ 5, 498–507 (2013). CAS PubMed PubMed Central Google Scholar * Sun, X. et al. PROTACs:
great opportunities for academia and industry. _Signal Transduct. Target. Ther._ 4, 64 (2019). PubMed PubMed Central Google Scholar * Cheng, M. et al. Discovery of potent and selective
epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. _J. Med. Chem._ 63, 1216–1232 (2020). CAS PubMed PubMed Central Google Scholar * Gu, S. et al. PROTACs: an
emerging targeting technique for protein degradation in drug discovery. _BioEssays_ 40, e1700247 (2018). PubMed Google Scholar * Leung, E. L. et al. Targeting tyrosine kinase
inhibitor-resistant non-small cell lung cancer by inducing epidermal growth factor receptor degradation via methionine 790 oxidation. _Antioxid. Redox Signal._ 24, 263–279 (2016). CAS
PubMed PubMed Central Google Scholar * Lee, J. Y. et al. Curcumin induces EGFR degradation in lung adenocarcinoma and modulates p38 activation in intestine: the versatile adjuvant for
gefitinib therapy. _PLoS ONE_ 6, e23756 (2011). CAS PubMed PubMed Central Google Scholar * Na, Y. S. et al. YM155 induces EGFR suppression in pancreatic cancer cells. _PLoS ONE_ 7,
e38625 (2012). CAS PubMed PubMed Central Google Scholar * Mao, J. et al. Arsenic circumvents the gefitinib resistance by binding to P62 and mediating autophagic degradation of EGFR in
non-small cell lung cancer. _Cell Death Dis._ 9, 963 (2018). PubMed PubMed Central Google Scholar * Xu, S. W. et al. Autophagic degradation of epidermal growth factor receptor in
gefitinib-resistant lung cancer by celastrol. _Int. J. Oncol._ 49, 1576–1588 (2016). CAS PubMed Google Scholar * Menard, L., Floc’h, N., Martin, M. J. & Cross, D. A. E. Reactivation
of mutant-EGFR degradation through clathrin inhibition overcomes resistance to EGFR tyrosine kinase inhibitors. _Cancer Res._ 78, 3267–3279 (2018). CAS PubMed Google Scholar * Lan, P. et
al. Synthesis and antiproliferative evaluation of 23-hydroxybetulinic acid derivatives. _Eur. J. Med. Chem._ 46, 2490–2502 (2011). CAS PubMed Google Scholar * Tan, X., Lambert, P. F.,
Rapraeger, A. C. & Anderson, R. A. Stress-induced EGFR trafficking: mechanisms, functions, and therapeutic implications. _Trends Cell Biol._ 26, 352–366 (2016). CAS PubMed PubMed
Central Google Scholar * Tomas, A., Futter, C. E. & Eden, E. R. EGF receptor trafficking: consequences for signaling and cancer. _Trends Cell Biol._ 24, 26–34 (2014). CAS PubMed
PubMed Central Google Scholar * Heukers, R. et al. Endocytosis of EGFR requires its kinase activity and N-terminal transmembrane dimerization motif. _J. Cell Sci._ 126, 4900–4912 (2013).
CAS PubMed Google Scholar * Tong, J. et al. Epidermal growth factor receptor phosphorylation sites Ser991 and Tyr998 are implicated in the regulation of receptor endocytosis and
phosphorylations at Ser1039 and Thr1041. _Mol. Cell Proteom._ 8, 2131–2144 (2009). CAS Google Scholar * Tanaka, T. et al. Ligand-activated epidermal growth factor receptor (EGFR) signaling
governs endocytic trafficking of unliganded receptor monomers by non-canonical phosphorylation. _J. Biol. Chem._ 293, 2288–2301 (2018). CAS PubMed Google Scholar * Sigismund, S. et al.
Clathrin-independent endocytosis of ubiquitinated cargos. _Proc. Natl. Acad. Sci. USA_ 102, 2760–2765 (2005). CAS PubMed PubMed Central Google Scholar * El-Sayed, A. & Harashima, H.
Endocytosis of gene delivery vectors: from clathrin-dependent to lipid raft-mediated endocytosis. _Mol. Ther._ 21, 1118–1130 (2013). CAS PubMed PubMed Central Google Scholar * Otto, G.
P. & Nichols, B. J. The roles of flotillin microdomains—endocytosis and beyond. _J. Cell Sci._ 124, 3933 (2011). CAS PubMed Google Scholar * Burslem, G. M. et al. The advantages of
targeted protein degradation over inhibition: an RTK case study. _Cell Chem. Biol._ 25, 67–77 e63 (2018). CAS PubMed Google Scholar * Sakanyan, V. et al. Screening and discovery of
nitro-benzoxadiazole compounds activating epidermal growth factor receptor (EGFR) in cancer cells. _Sci. Rep._ 4, 3977 (2014). PubMed PubMed Central Google Scholar * Iradyan, M. et al.
Targeting degradation of EGFR through the allosteric site leads to cancer cell detachment-promoted death. _Cancers_ 11, 1094 (2019). CAS PubMed Central Google Scholar * Sharma, S. V.,
Bell, D. W., Settleman, J. & Haber, D. A. Epidermal growth factor receptor mutations in lung cancer. _Nat. Rev. Cancer_ 7, 169–181 (2007). CAS PubMed Google Scholar * Huang, K. Y. et
al. Small molecule T315 promotes casitas B-lineage lymphoma-dependent degradation of epidermal growth factor receptor via Y1045 autophosphorylation. _Am. J. Respir. Crit. Care Med._ 193,
753–766 (2016). CAS PubMed Google Scholar * Alwan, H. A., van Zoelen, E. J. & van Leeuwen, J. E. Ligand-induced lysosomal epidermal growth factor receptor (EGFR) degradation is
preceded by proteasome-dependent EGFR de-ubiquitination. _J. Biol. Chem._ 278, 35781–35790 (2003). CAS PubMed Google Scholar * Sigismund, S., Avanzato, D. & Lanzetti, L. Emerging
functions of the EGFR in cancer. _Mol. Oncol._ 12, 3–20 (2018). PubMed Google Scholar * Thomas, R. & Weihua, Z. Rethink of EGFR in cancer with its kinase independent function on board.
_Front. Oncol._ 9, 800 (2019). PubMed PubMed Central Google Scholar * Ahsan, A. et al. Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors.
_Neoplasia (New York, N.Y.)_ 14, 670–677 (2012). CAS Google Scholar * Uemura, T., Kametaka, S. & Waguri, S. GGA2 interacts with EGFR cytoplasmic domain to stabilize the receptor
expression and promote cell growth. _Sci. Rep._ 8, 1368 (2018). PubMed PubMed Central Google Scholar * Zhao, X. C. et al. Systematic identification of CDC34 that functions to stabilize
EGFR and promote lung carcinogenesis. _EBioMedicine_ 53, 102689 (2020). PubMed PubMed Central Google Scholar * Kitai, Y. et al. STAP-2 protein promotes prostate cancer growth by enhancing
epidermal growth factor receptor stabilization. _J. Biol. Chem._ 292, 19392–19399 (2017). CAS PubMed PubMed Central Google Scholar * Ahsan, A. et al. Destabilization of the epidermal
growth factor receptor (EGFR) by a peptide that inhibits EGFR binding to heat shock protein 90 and receptor dimerization. _J. Biol. Chem._ 288, 26879–26886 (2013). CAS PubMed PubMed
Central Google Scholar * Sigismund, S. et al. Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. _Dev. Cell_ 15, 209–219 (2008).
CAS PubMed Google Scholar * Tan, X., Thapa, N., Sun, Y. & Anderson, R. A. A kinase-independent role for EGF receptor in autophagy initiation. _Cell_ 160, 145–160 (2015). CAS PubMed
PubMed Central Google Scholar * Zwang, Y. & Yarden, Y. p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. _EMBO J._ 25, 4195–4206
(2006). CAS PubMed PubMed Central Google Scholar * Tomas, A. et al. WASH and Tsg101/ALIX-dependent diversion of stress-internalized EGFR from the canonical endocytic pathway. _Nat.
Commun._ 6, 7324 (2015). CAS PubMed Google Scholar * Liao, H. J. & Carpenter, G. Cetuximab/C225-induced intracellular trafficking of epidermal growth factor receptor. _Cancer Res._
69, 6179–6183 (2009). CAS PubMed PubMed Central Google Scholar * Brand, T. M., Iida, M. & Wheeler, D. L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab.
_Cancer Biol. Ther._ 11, 777–792 (2011). CAS PubMed PubMed Central Google Scholar * Jaramillo, M. L. et al. Effect of the anti-receptor ligand-blocking 225 monoclonal antibody on EGF
receptor endocytosis and sorting. _Exp. Cell Res._ 312, 2778–2790 (2006). CAS PubMed Google Scholar * Oksvold, M. P., Pedersen, N. M., Forfang, L. & Smeland, E. B. Effect of
cycloheximide on epidermal growth factor receptor trafficking and signaling. _FEBS Lett._ 586, 3575–3581 (2012). CAS PubMed Google Scholar * Tanaka, T. et al. Ligand-activated epidermal
growth factor receptor (EGFR) signaling governs endocytic trafficking of unliganded receptor monomers by non-canonical phosphorylation. _J. Biol. Chem._ 293, 2288–2301 (2018). CAS PubMed
Google Scholar * Yao, N. et al. A piperazidine derivative of 23-hydroxy betulinic acid induces a mitochondria-derived ROS burst to trigger apoptotic cell death in hepatocellular carcinoma
cells. _J. Exp. Clin. Cancer Res._ 35, 192 (2016). PubMed PubMed Central Google Scholar * Yao, N. et al. Inhibition of PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant
cancer cells to B5G1, a new betulinic acid analog. _Cell Death Dis._ 10, 232 (2019). PubMed PubMed Central Google Scholar * Zhang, D. M. et al. BBA, a derivative of 23-hydroxybetulinic
acid, potently reverses ABCB1-mediated drug resistance in vitro and in vivo. _Mol. Pharm._ 9, 3147–3159 (2012). CAS PubMed PubMed Central Google Scholar * Ji, Z. N., Ye, W. C., Liu, G.
G. & Hsiao, W. L. 23-Hydroxybetulinic acid-mediated apoptosis is accompanied by decreases in bcl-2 expression and telomerase activity in HL-60 Cells. _Life Sci._ 72, 1–9 (2002). CAS
PubMed Google Scholar * Salazar, G. & Gonzalez, A. Novel mechanism for regulation of epidermal growth factor receptor endocytosis revealed by protein kinase A inhibition. _Mol. Biol.
Cell_ 13, 1677–1693 (2002). CAS PubMed PubMed Central Google Scholar * Wang, J. & Yu, R. K. Interaction of ganglioside GD3 with an EGF receptor sustains the self-renewal ability of
mouse neural stem cells in vitro. _Proc. Natl. Acad. Sci. USA_ 110, 19137–19142 (2013). CAS PubMed PubMed Central Google Scholar * Foerster, S. et al. Characterization of the EGFR
interactome reveals associated protein complex networks and intracellular receptor dynamics. _Proteomics_ 13, 3131–3144 (2013). CAS PubMed Google Scholar * Pan, S. et al. Target
identification of natural products and bioactive compounds using affinity-based probes. _Nat. Prod. Rep._ 33, 612–620 (2016). CAS PubMed Google Scholar * Odintsova, E., Voortman, J.,
Gilbert, E. & Berditchevski, F. Tetraspanin CD82 regulates compartmentalisation and ligand-induced dimerization of EGFR. _J. Cell Sci._ 116, 4557–4566 (2003). CAS PubMed Google Scholar
Download references ACKNOWLEDGEMENTS We are grateful to Prof. Kwok-Pui Fung for kindly offering HepG2/ADM cells. This work was supported by National Key R&D Program of China
(2017YFC1703800), Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01Y036), National Science Foundation of China (81630095, 81973340, 81903634 and
81803566), National Science and Technology Major Project (2018ZX09711001-008-008), National High-level Personnel of Special Support Program (Dongmei Zhang), Natural Science Foundation of
Guangdong Province (2019A1515010144) and China Postdoctoral Science Foundation-funded project (2019T120793). AUTHOR INFORMATION Author notes * These authors contributed equally: Nan Yao,
Chen-Ran Wang AUTHORS AND AFFILIATIONS * College of Pharmacy, Jinan University, Guangzhou, China Nan Yao, Chen-Ran Wang, Ming-Qun Liu, Ying-Jie Li, Wei-Min Chen, Zheng-Qiu Li, Chun-Lin Fan,
Min-Feng Chen, Ming Qi, Xiao-Bo Li, Dong-Mei Zhang & Wen-Cai Ye * Guangdong Province Key Laboratory of Pharmacodynamic Constituents of Traditional Chinese Medicine and New Drugs
Research, Jinan University, Guangzhou, China Nan Yao, Chen-Ran Wang, Chun-Lin Fan, Min-Feng Chen, Ming Qi, Xiao-Bo Li, Dong-Mei Zhang & Wen-Cai Ye * School of Medicine, Jinan University,
Guangzhou, China Qi Qi & Jian Hong * State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macao, China Jin-Jian Lu
Authors * Nan Yao View author publications You can also search for this author inPubMed Google Scholar * Chen-Ran Wang View author publications You can also search for this author inPubMed
Google Scholar * Ming-Qun Liu View author publications You can also search for this author inPubMed Google Scholar * Ying-Jie Li View author publications You can also search for this author
inPubMed Google Scholar * Wei-Min Chen View author publications You can also search for this author inPubMed Google Scholar * Zheng-Qiu Li View author publications You can also search for
this author inPubMed Google Scholar * Qi Qi View author publications You can also search for this author inPubMed Google Scholar * Jin-Jian Lu View author publications You can also search
for this author inPubMed Google Scholar * Chun-Lin Fan View author publications You can also search for this author inPubMed Google Scholar * Min-Feng Chen View author publications You can
also search for this author inPubMed Google Scholar * Ming Qi View author publications You can also search for this author inPubMed Google Scholar * Xiao-Bo Li View author publications You
can also search for this author inPubMed Google Scholar * Jian Hong View author publications You can also search for this author inPubMed Google Scholar * Dong-Mei Zhang View author
publications You can also search for this author inPubMed Google Scholar * Wen-Cai Ye View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
Conception and design of study: N.Y., D.-M.Z. and W.-C.Y. Acquisition of data: N.Y. and C.-R.W. (all experiment), M.-Q.L. (Western blot), M.-F.C., Y.-J.L., M.Q. and X.-B.L. (animal study).
Analysis and interpretation of data: N.Y., C.-R.W., J.-J.L., Q.Q., D.-M.Z. and W.-C.Y. Compounds preparation: Z.-Q.L. (synthesis of DPBA-1), W.-M.C. and C.-L.F. (synthesis of DPBA). Writing
of the manuscript: N.Y., D.-M.Z. and W.-C.Y. Revision of the manuscript: N.Y., Q.Q., J.-J.L., J.H., D.-M.Z. and W.-C.Y. CORRESPONDING AUTHORS Correspondence to Dong-Mei Zhang or Wen-Cai Ye.
ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURES SUPPLEMENTARY MATERIALS SUPPLEMENTARY INFORMATION RIGHTS
AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in
any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Yao, N., Wang,
CR., Liu, MQ. _et al._ Discovery of a novel EGFR ligand DPBA that degrades EGFR and suppresses EGFR-positive NSCLC growth. _Sig Transduct Target Ther_ 5, 214 (2020).
https://doi.org/10.1038/s41392-020-00251-2 Download citation * Received: 07 April 2020 * Revised: 25 May 2020 * Accepted: 15 June 2020 * Published: 09 October 2020 * DOI:
https://doi.org/10.1038/s41392-020-00251-2 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative