Methyl-cpg-binding protein 2 drives the furin/tgf-β1/smad axis to promote epithelial–mesenchymal transition in pancreatic cancer cells
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Methyl-CpG-binding protein 2 (MeCP2) has been characterized as an oncogene in several types of cancer. However, its precise role in pancreatic ductal adenocarcinoma (PDAC) remains
unclear. Hence, this study aimed to evaluate the potential role of MeCP2 in pancreatic cancer progression. We found that MeCP2 was upregulated in pancreatic cancer tissues, enhanced
migration, invasion, and proliferation in pancreatic cancer cells, and promoted tumorigenesis. Further evidence revealed that MeCP2 remarkably increased the mesenchymal markers vimentin,
N-cadherin, and Snail, and downregulated the expression of the epithelial markers E-cadherin and ZO-1, indicating that MeCP2 promotes epithelial–mesenchymal transition (EMT). In addition, we
found that MeCP2 upregulated the expression of Furin, activated TGF-β1, and increased the levels of p-Smad2/3. Importantly, we demonstrated that MeCP2, as a coactivator, enhanced Smad3
binding to the _furin_ promoter to improve its transcription. Therefore, MeCP2/Smads drive the expression of Furin to activate TGF-β1, and in turn, phosphorylate Smad2/3, which forms a
positive-feedback axis to promote EMT in pancreatic cancer cells. SIMILAR CONTENT BEING VIEWED BY OTHERS S100A2 INDUCES EPITHELIAL–MESENCHYMAL TRANSITION AND METASTASIS IN PANCREATIC CANCER
BY COORDINATING TRANSFORMING GROWTH FACTOR Β SIGNALING IN SMAD4-DEPENDENT MANNER Article Open access 27 September 2023 PRMT5 METHYLATING SMAD4 ACTIVATES TGF-Β SIGNALING AND PROMOTES
COLORECTAL CANCER METASTASIS Article 29 March 2023 NSUN2 STIMULATES TUMOR PROGRESSION VIA ENHANCING _TIAM2_ MRNA STABILITY IN PANCREATIC CANCER Article Open access 01 July 2023 INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC), the most common type of pancreatic cancer, is an extremely lethal disease characterized by mortality closely reflecting incidence. Because of the
absence of reliable early biomarkers and the lack of efficient therapies, the 5-year survival rate remains below 9%1. Despite decades of effort, the underlying mechanisms of pancreatic
cancer remain far from fully understood. Thus, there is an urgent need to discover the mechanism of pancreatic cancer to facilitate the prediction of cancer risk and diagnose this disease at
an earlier stage. Epithelial–mesenchymal transition (EMT) is a critical step at several stages during tumor progression, that is characterized by a breakdown of cell junctions and the loss
of epithelial characteristics and cell polarity, leading to cancer progression2. Moreover, it has been reported that EMT plays a significant role in the migration and invasion of various
malignant cells, including pancreatic cancer cells. Meanwhile, it is generally accepted that the activation of TGF-β1/Smad signaling is critical to induce EMT3,4. Furin has been reported to
promote the migration and invasion abilities of human cancer cells via pro-MT1-MMP and/or pro-TGF-β1 activation5,6. Furin is a member of the family of subtilisin/kexin-like proprotein
convertases (PCs), which contribute to the activation of precursor pro-proteins to become biologically active7. Methyl-CpG-binding protein 2 (MeCP2) is a key member of the methyl-CpG-binding
domain proteins (MBDs), which are important components of the DNA methylation machinery8. The classical model of MeCP2 function delineates its role in gene suppression through binding to
methylated CpG dinucleotides in promoters. Nevertheless, recent discoveries have proven that MeCP2 is a multifunctional protein involved not only in transcriptional silencing but also in
transcriptional activation, chromatin compaction, and modulation of RNA splicing9. The hypothesis has especially been focused on MeCP2 acting as a transcriptional activator to stimulate gene
expression, which has been confirmed in the hypothalamus10. Recently, it has been found that MeCP2 is involved in the development of tumors in different cancers, such as gastric
carcinoma11, cervical cancer12, and prostate cancer13. However, the role of MeCP2 in pancreatic cancer remains to be clarified. In this study, we demonstrated that MeCP2 was markedly
different in pancreatic cancer tissues, enhanced the proliferation, migration, and invasion abilities of pancreatic cancer cells, and promoted tumorigenesis. Furthermore, it was found that
MeCP2, as a coactivator, enhanced Smad3 binding to the _furin_ promoter to activate Furin/ TGF-β1/Smad signaling resulting in the promotion of EMT in pancreatic cancer cells. All these
findings prove for the first time that MeCP2 might be a promoter in pancreatic cancer progression. RESULTS MECP2 IS PROFILED IN PANCREATIC CANCERS AND DIFFERENT PANCREATIC CANCER CELLS To
confirm the clinical relevance of MeCP2 expression, we first analyzed MeCP2 mRNA expression in the Badea pancreas database. We found that the MeCP2 mRNA level was higher in pancreatic cancer
tissues than in normal pancreatic tissues (1.724 ± 0.05294 vs. 1.431 ± 0.07816, _P_ < 0.01, _n_ = 78) (Fig. 1a). To evaluate the correlation of MeCP2 expression and clinicopathological
features, we further interrogated the TCGA database (https://genome-cancer.ucsc.edu). We analyzed the mRNA level of MeCP2 at different stages and found that MeCP2 expression was higher in
stage II–IV groups than in the stage I group (Fig. 1b, c). Next, we assessed the expression of MeCP2 in three pancreatic cancer cell lines, PANC1, SW1990, and PaTu8988, using real-time PCR
and western blotting. The results showed that MeCP2 mRNA and protein levels were higher in PANC1 cells than in SW1990 and PaTu8988 cells (Fig. 1d, e). In addition, we found that PANC1 cells
had poorer differentiation and were more invasive compared to the other two cell lines (Supplementary Fig. S1a–c), indicating that MeCP2 was associated with the state of differentiation or
the malignant phenotype of these cells. Subsequently, we constructed shMeCP2 and Flag-MeCP2 plasmids to investigate the roles of MeCP2 in pancreatic cancer, with shEGFP or vector as a
control, respectively. In previous study, we found that MeCP2 expression, at both the mRNA and protein levels, was highest in PANC1 cells, intermediate in PaTu8988 cells, and lowest in
SW1990 cells. Therefore, we used PANC1 and PaTu8988 cells with higher MeCP2 expression in shRNA experiments and SW1990 and PaTu8988 cells with relatively lower MeCP2 expression in MeCP2
overexpression assays. After infection, the mRNA and protein levels of MeCP2 were significantly reduced in the shMeCP2 group compared with the shEGFP group (Supplementary Fig. S1d, e).
Vector or Flag-MeCP2 was transfected into PaTu8988 and SW1990 cells, and MeCP2 overexpression was confirmed at the mRNA and protein levels by real-time PCR and western blotting
(Supplementary Fig. S1f, g). MECP2 PROMOTES PROLIFERATION OF PANCREATIC CANCER CELLS AND TUMORIGENESIS To determine the role of MeCP2 in the proliferation of pancreatic cancer cells, we used
the CCK8 assay to generate growth curves. We found that MeCP2 knockdown caused a lower proliferation of PANC1 and PaTu8988 cells at 2, 3, 4, and 5 days after infection (Fig. 2a, b).
Similarly, MeCP2 overexpression enhanced the cell proliferation ability at 2, 3, 4, and 5 days in SW1990 and PaTu8988 cells (Fig. 2c, d). Furthermore, experiments in the xenograft mouse
model proved that MeCP2 also significantly increased tumor growth (Fig. 2e–h). These results suggest that MeCP2 is critical for cell proliferation in pancreatic cancer cells. MECP2 ENHANCES
THE ABILITY OF MIGRATION AND INVASION IN PANCREATIC CANCER CELLS Next, we examined the migration ability of pancreatic cancer cells by transwell assays. We found that the numbers of migrated
cells were 154 ± 14 and 56 ± 9 (_P_ < 0.05) in shEGFP and shMeCP2 PANC1 cells, and 120 ± 12 and 45 ± 8 (_P_ < 0.01) in shEGFP and shMeCP2 PaTu8988 cells, respectively (Fig. 3a, b),
indicating that MeCP2 knockdown significantly decreased their migration ability. To confirm the above results, we assessed the migration ability of PaTu8988 cells and SW1990 cells
transfected with the vector or the Flag-MeCP2 plasmid. The numbers of migrated cells were 39 ± 6 and 80 ± 10 (_P_ < 0.05) in vector and Flag-MeCP2 SW1990 cells, and 114 ± 12 and 226 ± 16
(_P_ < 0.01) in vector and Flag-MeCP2 PaTu8988 cells, respectively (Fig. 3c, d), indicating that upregulation of MeCP2 significantly increased their migration ability. All these data
suggest that MeCP2 enhances the migration ability of pancreatic cancer cells. Subsequently, we examined the invasion ability by BD Matrigel invasion assays. PANC1 and PaTu8988 cells were
transfected with the shEGFP or shMeCP2 plasmid, and PaTu8988 and SW1990 cells were transfected with the vector or Flag-MeCP2 for 72 h. The numbers of invasive cells were 159 ± 12 and 78 ± 12
(_P_ < 0.05) in shEGFP and shMeCP2 PANC1 cells, and 116 ± 11 and 48 ± 10 (_P_ < 0.01) in shEGFP and shMeCP2 PaTu8988 cells, respectively (Fig. 3e, f), indicating that MeCP2 knockdown
obviously suppressed the invasion ability of PANC1 and PaTu8988 cells. In addition, the numbers of invasive cells were 42 ± 8 and 105 ± 11 (_P_ < 0.01) in vector and Flag-MeCP2 SW1990
cells, and 122 ± 12 and 188 ± 12 (_P_ < 0.001) in vector and Flag-MeCP2 PaTu8988 cells, respectively (Fig. 3g, h), suggesting that upregulation of MeCP2 significantly enhanced the
invasion ability of PaTu8988 and SW1990 cells. In addition, MeCP2 knockdown inhibited the expression of MMP2 and MMP9 (pro- and total) (Fig. 3i and Supplementary Fig. S2a, b, e, f), while
MeCP2 overexpression resulted in upregulation of MMP2 and MMP9 (pro- and total) (Fig. 3j and Supplementary Fig. S2c, d, g, h). All these findings indicate that MeCP2 enhances the invasion
ability of these three kinds of pancreatic cancer cells. MECP2 DRIVES TGF-Β1/SMAD SIGNALING TO PROMOTE EMT IN PANCREATIC CANCER CELLS A previous study showed that EMT is associated with
migration and invasion in cancer cells14. Therefore, we detected EMT markers at the protein level using western blotting. Our results revealed that MeCP2 knockdown induced upregulation of
E-cadherin and ZO-1 and downregulation of vimentin, N-cadherin, and Snail (Fig. 4a). Conversely, MeCP2 overexpression resulted in downregulation of E-cadherin and ZO-1 and upregulation of
vimentin, N-cadherin, and Snail (Fig. 4b). In addition, we found that the cells acquired a spindle-shaped morphology characteristic of EMT15 after overexpression of MeCP2 in PANC1, PaTu8988,
and SW1990 cells (Supplementary Fig. S3a). Using immunofluorescence studies, we found that MeCP2 could promote vimentin and N-cadherin while inhibiting E-cadherin (Supplementary Fig.
S4a–f). To further examine the mechanism by which MeCP2 affects EMT in pancreatic cancer cells, we investigated the effects of MeCP2 on classic TGF-β1/Smad signaling. The results showed that
MeCP2 knockdown downregulated active TGF-β1 and p-Smad2/3 (Fig. 4c), while MeCP2 overexpression resulted in the upregulation of active TGF-β1 and p-Smad2/3 (Fig. 4d). MeCP2 knockdown or
overexpression had no influence on total Smad2/3, total TGF-β1, or TGF-βR expression (Fig. 4c–f). In addition, we found that MeCP2 stimulated the activation of TGF-β1 in the supernatant
(Supplementary Fig. S5a, b) and hardly affected the expression of TGF-β2 and TGF-β3 (Supplementary Fig. S5c, d). All these findings indicate that MeCP2 drives active TGF-β1/Smad signaling to
promote EMT in pancreatic cancer cells. EXOGENOUS TGF-Β1 ENHANCES MIGRATION, INVASION, PROLIFERATION, AND SMAD2/3 PHOSPHORYLATION IN PANCREATIC CANCER CELLS To confirm whether pancreatic
cancer cells are TGF-β1 responsive, we determined the effects of exogenous TGF-β1 on migration, invasion, and Smad2/3 phosphorylation in pancreatic cancer cells. We found that exogenous
TGF-β1 significantly promoted the migration and invasion abilities of PaTu8988, PANC1, and SW1990 cells (Fig. 5a–i). In addition, we found that exogenous TGF-β1 promoted the growth of
PaTu8988 and SW1990 cells (Supplementary Fig. S3b, c). Moreover, exogenous TGF-β1 enhanced Smad2/3 phosphorylation in these three cell lines (Fig. 5j). All these findings suggest that
exogenous TGF-β1 can mimic the effect of MeCP2 overexpression on migration, invasion, and Smad phosphorylation in the three cell lines. MECP2 REGULATES THE EXPRESSION OF FURIN TO ACTIVATE
TGF-Β1 TGF-β1 is deemed to be processed by the subtilisin-like proprotein convertase, Furin5. In this study, we found that exogenous TGF-β1 could induce Furin expression at both the mRNA and
protein levels (Supplementary Fig. S3d, e). To further confirm the role of MeCP2 in the regulation of Furin, we transfected PANC1 and PaTu8988 cells with shEGFP or shMeCP2, and SW1990 and
PaTu8988 cells with vector or Flag-MeCP2. As expected, MeCP2 knockdown downregulated Furin expression at the mRNA and protein levels compared with shEGFP expression (Fig. 6a, b).
Accordingly, MeCP2 overexpression resulted in upregulation of Furin expression at the mRNA and protein levels (Fig. 6c, d). Further results showed that Furin knockdown or inhibitor decreased
the expression of active TGF-β1 (Fig. 6e, f). In addition, after we transfected PaTu8988 and SW1990 cells with Flag-MeCP2, Furin knockdown downregulated active TGF-β1 and p-Smad2/3 (Fig.
6g), indicating that knockdown of Furin could blunt the MeCP2 overexpression effects. Conversely, Furin overexpression upregulated active TGF-β1 and p-Smad2/3 after we infected PANC1 and
PaTu8988 cells with shMeCP2 virus suspension (Fig. 6h), indicating that overexpression of Furin could rescue the MeCP2 knockdown effects. All these results suggest that MeCP2 plays an
important role in regulating Furin expression to enhance active TGF-β1. MECP2 BINDS TO SMAD3 TO PROMOTE FURIN TRANSCRIPTION To characterize the interaction between MeCP2 and Smad2, Smad3,
and Smad4, we first used immunoprecipitation assay to detect the interaction between MeCP2 and Smad2, Smad3, and Smad4. The results revealed that MeCP2 bound to Smad2, Smad3, and Smad4 (Fig.
7a–c). Then, we performed Chromatin immunoprecipitation (ChIP) assays to examine the binding ability of Smad2, Smad3, Smad4, and MeCP2 to three potential transcriptional binding sites on
the _furin_ promoter (Fig. 7d–j). Our data showed that Smad3 could bind to the promoter of three potential transcriptional binding sites of _furin_ (Δ-1674-Δ-1662, Δ-1125-Δ-1113, and
Δ-764-Δ-752), Smad2 could bind to site 1 (Δ-1674-Δ-1662), and site 2 (Δ-1125-Δ-1113) and Smad4 could only bind to site 2 (Fig. 7e–g), while MeCP2 could not bind to the promoter (Fig. 7h–j).
Transcription factor-binding sites that are located closer to translational start sites are more relevant to gene transcriptional activity16. It has been suggested that Smad3 may have more
influence on _furin_ transcription than Smad2/4. In addition, we found that knockdown of MeCP2 could weaken the ability of Smad2/3/4 to bind to the _furin_ promoter (Supplementary Fig.
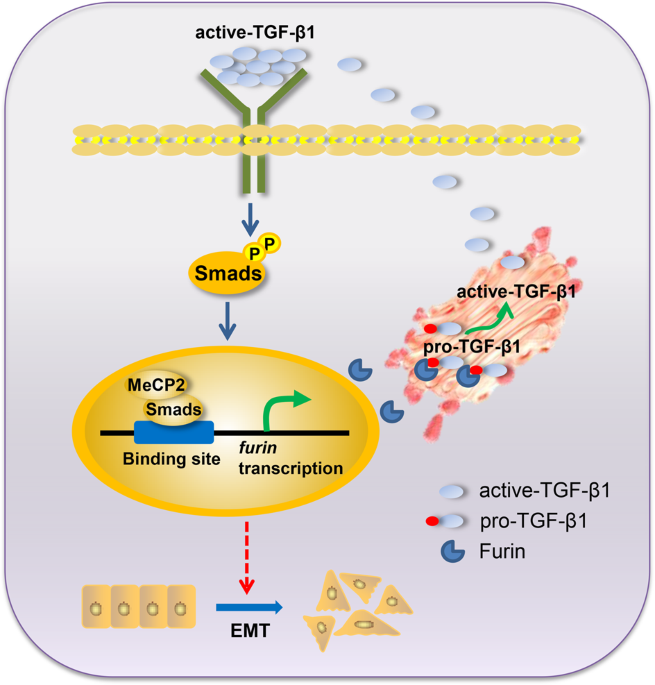
S5e–g). Thus, we proposed that Smad2/3/4, but mainly Smad3, bound to the _furin_ promoter by interacting with MeCP2, to enhance the transcription of _furin_. DISCUSSION The above results
indicate that MeCP2 may function as a promoter in pancreatic cancer. We confirmed that MeCP2 was upregulated in human pancreatic cancer and was directly related to clinicopathological
features and stage. Furthermore, we found for the first time that the MeCP2-driven Smads–Furin-TGF-β1 axis represents a novel mechanism for promoting EMT in pancreatic cancer cells. All
these findings suggest that MeCP2 may be a potential candidate for the diagnosis of pancreatic cancer. Ever since the discovery that MeCP2 is an essential player in Rett syndrome (RTT),
there has been considerable interest in obtaining a comprehensive understanding of this protein. However, the involvement of MeCP2 in pathologies other than RTT, such as tumorigenesis,
remains poorly explored and understood. MeCP2 is upregulated in gastric, breast, colon, and prostate cancer9. In gastric cancer cells, MeCP2 was found to promote proliferation by activation
of the MEK1/2–ERK1/2 signaling pathway through upregulating GIT112. Yadav et al.17 identified MeCP2 gene polymorphisms as candidates for breast cancer susceptibility, while Kedarlal Sharma
et al.18 proved that MeCP2 overexpression inhibited the proliferation, migration, and invasion of C6 glioma cells. Nevertheless, to our knowledge, few studies have described the relationship
between MeCP2 and EMT in pancreatic cancer cells. It is well-known that EMT plays an important role in pancreatic carcinoma progression19. In this study, we report that MeCP2 promotes EMT
by driving Furin/TGF-β1/Smad signaling in pancreatic cancer cells. TGF-β1 signaling is associated with the regulation of malignancy initiation, progression, and metastasis in mammary
carcinoma, pancreatic cancer, glioblastoma, prostate carcinoma, and hepatocellular carcinoma20. When TGF-β1 is activated, Smad2 and Smad3 are phosphorylated and undergo dimerization with
Smad4, thus allowing its translocation into the nucleus21. As expected, MeCP2 knockdown downregulated active TGF-β1 and p-Smad2/3, while MeCP2 overexpression upregulated active TGF-β1, and
then activated p-Smad2/3, suggesting that MeCP2 activates TGF-β1/Smad signaling to regulate EMT. The classical role of MeCP2 is in gene suppression through recruitment of histone
deacetylases and co-repressor complexes to methylated CpG-sites9, which has been observed in numerous diseases, including Rett syndrome22, scleroderma fibroblasts23 and systemic sclerosis24.
However, in recent studies, MeCP2 has been found to play an important role in transcription activation25. After studying thousands of genes, the majority of genes (~85%), such as Sst,
Oprk1, Gamt, and Gprin117, appear to be activated by MeCP225. Moreover, Furin is an authentic TGF-β1-converting enzyme and thus increases activated TGF-β1 levels25. In this study, we found a
positive correlation between MeCP2 and Furin expression and confirmed that MeCP2 enhances Smad2/3/4, especially Smad3 binding to the _furin_ promoter. Notably, our results establish a
direct link between MeCP2, Smad3, and Furin in controlling EMT in pancreatic cancer cells. In summary, our study provides the first evidence that MeCP2, Smad3, and Furin form positive
feedback to promote EMT in pancreatic cancer cells. All these findings suggest that MeCP2 is a potential biomarker for pancreatic cancer therapy. MATERIALS AND METHODS ANALYSIS OF MECP2 MRNA
EXPRESSION IN HUMAN PANCREATIC CANCER TISSUES Correlations between pancreatic cancer histology and MeCP2 gene expression were determined through analysis of Badea and TCGA databases, which
are available through Oncomine (Compendia Biosciences, www.oncomine.org) and UCSC (https://genome-cancer.ucsc.edu). IMMUNOHISTOCHEMICAL STAINING Seventy-eight pancreatic cancer tissues from
Shanghai Outdo Biotech Company (NO.XT15-051) which were got patient informed consent and approved by the China Association For Ethical Studies were enrolled in this study. In this study,
immunohistochemistry (IHC) was performed on tissue-microarrays. IHC was done following the standard protocol of Peroxidase Conjugated Mouse/Rabbit IgG SABC Kit (Catalog No: SA1020, Boster
Biological Technology). The primary antibody was a rabbit monoclonal antibody against MeCP2 (Cell Signaling, CAT 3456, 1:500), and the secondary antibody was biotinylated goat
anti-mouse/anti-rabbit IgG (Boster Biological technology, BA1056, 1:200). The chromogenic reaction was carried out with a DAB chromogenic kit (Catalog No: AR1022, Boster Biological
technology) for 5 min, resulting in the expected brown-colored signal. Finally, after rinsing with deionized water, the slides were counterstained with hematoxylin, dehydrated, and mounted
with mounting medium (Product No: D0547, Boster Biological technology) and coverslip. The colored cells are positive cells. CELL CULTURE The human pancreatic cancer cell lines (PANC1,
PaTu8988 and SW1990) were bought from Shanghai Institutes for Biological Sciences (CAS), and the human embryonic kidney cell line (293T) was obtained from the American Type Culture
Collection. All cell lines have been tested and authenticated by STR karyotype analysis. The cell lines were cultured with DMEM (Hyclone, China) supplemented with 10% fetal bovine serum
(Gibco, USA) in humidified 5% CO2 incubator at 37 °C. PLASMID CONSTRUCTION The entire MeCP2 sequence was amplified with RT-PCR using primers MeCP2-all (Table 1), and then cloned into the
expression vector p3xFLAG-Myc-CMV™-24 (Sigma, E9283). The MeCP2, EGFP shRNA oligos (Table 1) were firstly annealed into double-strands and then cloned into pLKO.1-puro (Sigma, SHC005).
TRANSFECTION OF THE PLASMID, VIRAL INFECTION, AND FURIN INHIBITORS In total, 5 × 105 PaTu8988 and SW1990 cells were seeded in a six-well plate per well for 12 h followed by transfection with
a vector or Flag-MeCP2 via Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA), in the light of the manufacturer’s instructions. In HEK293T cells, the plasmid shEGFP or sh- MeCP2 was
co-transfected with psPAX2 and pMD2.G, and the supernatants were harvested and concentrated following 48 h and 72 h. PANC1 and PaTu8988 cells were treated with 1 × 106 virus suspension of
either shEGFP or shMeCP2 in 8 mg/ml polybrene (Sigma-Aldrich) existence for 2 days. Cells were then cultivated with puromycin (1:10,000 dilution), while the cells in a blank group were all
unviable. Cells were incubated in culture medium with 10% FBS supplemented with Furin inhibitors Hexa-D-arginine (MCE, HY-P1028) over the concentration range 0, 0.1, and 1 mg/L. Cells were
harvested at 48 h after treatment. WESTERN BLOTTING The cultured cells were rinsed with cold PBS before treated with cell lysis buffer aids (2 × loading buffer, 2 µg/ml aprotinin, 1 mM PMSF,
2 mM β-mercaptoethanol) at 100 °C for 10 min. Then the mixture was centrifuged under 4 °C at 12000 rpm for 10 min. About 25–30 μg of protein was loaded in each lane, separated by 10%
SDS-PAGE, and transferred to the PVDF membrane. The membrane was blocked with 5% nonfat milk powder for 1 h at room temperature before overnight incubation with primary antibodies at 4 °C,
followed by the secondary antibody. The blots were incubated with primary antibodies rabbit anti-MeCP2 (Cell Signaling, CAT 3456), rabbit anti-Furin (Proteintech, CAT 18413-1-AP),
anti-TGF-β1 (Cell Signaling, CAT 3709), anti-TGF-βR (Cell Signaling, CAT 3712), anti-MMP2 (Immunoway, CAT YT2798), anti-MMP9 (Immunoway, CAT YT1892), EMT Kit (Cell Signaling, CAT 9782),
Smad2/3 Antibody Sampler Kit (Cell Signaling, CAT 12747), mouse anti-Flag (SIGMA, CAT 6631), mouse anti-β-Tubulin (Cell Signaling, CAT4466). CELL PROLIFERATION ASSAY Cell proliferation was
measured by Cell Counting Kit-8 (CCK8). Forty-eight hours later after transfection, cell suspension containing 1000–1500 cells (100 μL) was seeded into each well of a 96-well culture plate
and continued to incubate at 37 °C. At 1, 2, 3, 4, and 5 days, 100 μl serum-free culture medium and 10 μl CCK8 solutions were added to each well, followed by incubation at 37 °C for 2 h. The
absorbance was measured with a plate reader at 450 nm on an ELX-800 spectrometer reader. Six independent samples were detected in each experimental group. Cell proliferation rate =
(ODexperimental − ODblank)/(ODcontrol − ODblank) × 100%. XENOGRAFT MOUSE MODEL The protocol was approved by the Institutional Animal Care and Use Committee of Jiangsu University, Zhenjiang,
China. PANC1 and SW1990 cells (2.0 × 106 cells/site) stably transfected with shEGFP, shMeCP2, vector, and Flag-MeCP2 were subcutaneously injected into 5-week-old BALB/c nude mice (Shanghai
SLAC Laboratory Animal Co., Ltd, Shanghai, China) to generate xenografts. Five female mice were randomly distributed to each group. There is no blinding. The tumors were collected from the
fourth week. The tumor volume was measured every week after injection and calculated using the following formula: length × (width2)/2. TRANSWELL AND INVASION ASSAY Transwell and invasion
assays were carried out using matrigel chambers (BD Biosciences) according to the manufacturer’s protocol. Transfected PANC1, PaTu8988, and SW1990 cells were harvested and resuspended in
serum-free medium, cell suspensions containing 50,000 cells (100 μL) were plated into the upper chamber of the transwell, and DMEM (500 μl) containing 10% FBS was added to the lower chamber
of the transwell. The system was incubated at 37 °C, 5% CO2 atmosphere for 24 h. The cells on the upper surface were scraped and washed away, whereas the migrated and invaded cells on the
lower surface were fixed and stained with 0.05% crystal violet for 30 min. Finally, migrated and invaded cells were counted, and the relative number was calculated by software Image J.
REAL-TIME PCR The total RNA was isolated using RNAiso Plus (Takara). Reverse transcription was performed using RevertAid First Strand cDNA Synthesis Kit (Thermo) according to the
manufacturer’s recommendations. Real-time PCR was performed in triplicate in 20-μl reactions with iQ SYBR® Premix Ex Taq™ Perfect Real Time (Bio-Rad Laboratories, Inc.), 50 ng of
first-strand cDNA and 0.2 μg each primer. Samples were cycled once at 95 °C for 2 min, and then subjected to 35 cycles of 95, 56, and 72 °C for 30 s each. The relative mRNA content was
calculated using the 2−ΔΔCT method with GAPDH as an endogenous control. Primers are listed in Table 1. IMMUNOFLUORESCENCE Cells were seeded on glass coverslips in 24-well plates and grew to
subconfluence, then infected with shEGFP, shMeCP2, vector, or Flag-MeCP2 for 48 h. Routinely, cells were washed with PBS, fixed with ice-cold 100% methanol for 15 min at room temperature,
and blocked with 3% BSA in PBS for 1 h. Cells were incubated with primary antibody overnight at 4 °C, and then incubated with secondary antibodies for 1 h at room temperature in the dark,
DAPI (1 μg/ml, Pierce, IL, USA) counterstain was used for nuclear staining. After extensive washing, the coverslips were then mounted on glass slides, and the fluorescent images were
captured with a fluorescent microscope and a SPOT CCD camera. IMMUNOPRECIPITATION (IP) 293T cells were lysed in co-IP buffer (10 mM HEPES [pH 8.0], 300 mM NaCl, 0.1 mM EDTA, 20% glycerol,
0.2% NP-40, protease, and phosphatase inhibitors). Lysates were centrifuged and cleared by incubation with 25 μl of Protein A/G beads (Thermo Fisher Scientific, MA, USA) for 1 h at 4 °C. The
pre-cleared supernatant was subjected to IP using the indicated first antibodies at 4 °C overnight. Then, the protein complexes were collected by incubation with 30 μl of protein A/G beads
for 3–4 h at 4 °C. The collected protein complexes were washed with co-IP buffer and analyzed by western blotting. CHIP ANALYSIS ChIP was performed using Simple ChIP Plus Enzymatic chromatin
IP Kit (Agarose Beads) (Cell Signaling, CAT9004) according to the manufacturer’s recommendations. Cells were fixed with formaldehyde and lysed, and then chromatin was fragmented by partial
digestion with micrococcal nuclease automatic fragmentation to obtain chromatin fragments of 1–6 nucleosomes. Chromatin immunoprecipitations were performed using 1–2 μl of input, IgG, H3,
Smad2, Smad3, Smad4 antibodies, and ChIP-Grade Protein G Agarose Beads. After reversal of protein–DNA cross-links, the DNA was purified using DNA purification spin columns. CHIP-PCR
According to the online primer program (http://jaspar.genereg.net/), three potential transcriptional binding sites were predicted. Primers are listed in Table 1. The enrichment of particular
DNA sequences of PaTu8988 cells was analyzed by semi-quantitative PCR. Semi-quantitative PCR products were separated on 1.5% agarose gels, and visualized under UV illumination. The reaction
mixture contains 2 × green PCR mix 10 µl, 5 µM primers 1 µl, nuclease-free water 7 µl, and the appropriate DNA sample 2 µl. The 2% input chromatin DNA was diluted five times. Reactions were
carried out in a thermal cycler under the following conditions: 95 °C for 5 min followed by 95 °C for 30 s, 58 °C for 30 s, 72 °C for 30 s for 35 cycles, 72 °C for 5 min. DATA ANALYSIS All
data are presented as mean ± s.d. from at least three independent experiments. Comparisons between groups were analyzed using the Student’s _t_ test (two groups) or the one-way ANOVA
(multiple groups). _P_ < 0.05 was considered statistically significant. REFERENCES * Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2019. _CA Cancer J. Clin._ 69, 7–34
(2019). Google Scholar * Aiello, N. M. & Kang, Y. Context-dependent EMT programs in cancer metastasis. _J. Exp. Med._ 216, 1016–1026 (2019). Article CAS Google Scholar * Jiang, Y. et
al. Prosaposin is a biomarker of mesenchymal glioblastoma and regulates mesenchymal transition through the TGF-beta1/Smad signaling pathway. _J. Pathol._ 249, 26–38 (2019). Article CAS
Google Scholar * Zeng, K. et al. The pro-metastasis effect of circANKS1B in breast cancer. _Mol. Cancer_ 17, 160 (2018). Article CAS Google Scholar * Willson, J. A., Muir, C. A., Evered,
C. L., Cepeda, M. A. & Damjanovski, S. Stable expression of alpha1-antitrypsin Portland in MDA-MB-231 cells increased MT1-MMP and MMP-9 levels, but reduced tumour progression. _J. Cell
Commun. Signal_ 12, 479–488 (2018). Article CAS Google Scholar * Ventura, E., Weller, M. & Burghardt, I. Cutting edge: ERK1 mediates the autocrine positive feedback loop of TGF-beta
and furin in glioma-initiating cells. _J. Immunol_. 198, 4569–4574 (2017). * Jaaks, P. & Bernasconi, M. The proprotein convertase furin in tumour progression. _Int. J. Cancer_ 141,
654–663 (2017). Article CAS Google Scholar * Picard, N. & Fagiolini, M. MeCP2: an epigenetic regulator of critical periods. _Curr. Opin. Neurobiol._ 59, 95–101 (2019). Article CAS
Google Scholar * Pandey, S. & Pruitt, K. Functional assessment of MeCP2 in Rett syndrome and cancers of breast, colon, and prostate. _Biochem. Cell Biol._ 95, 368–378 (2017). Article
CAS Google Scholar * Chahrour, M. et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. _Science_ 320, 1224–1229 (2008). Article CAS Google
Scholar * Zhao, L. Y. et al. MeCP2, a target of miR-638, facilitates gastric cancer cell proliferation through activation of the MEK1/2-ERK1/2 signaling pathway by upregulating GIT1.
_Oncogenesis_ 6, e368 (2017). Article CAS Google Scholar * Pan, Z. X., Chen, S. R. & Li, C. Z. Upregulated exosomal miR-221/222 promotes cervical cancer via repressing
methyl-CpG-binding domain protein 2. _Eur. Rev. Med. Pharmacol. Sci_. 23, 3645–3653 (2019). * Guan, Y. et al. Epigenetic silencing of miR-137 induces resistance to bicalutamide by targeting
TRIM24 in prostate cancer cells. _Am. J. Transl. Res_. 11, 3226–3237 (2019). * Dongre, A. & Weinberg, R. A. New insights into the mechanisms of epithelial-mesenchymal transition and
implications for cancer. _Nat. Rev. Mol. Cell Biol._ 20, 69–84 (2019). Article CAS Google Scholar * Zheng, X. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but
induces chemoresistance in pancreatic cancer. _Nature_ 527, 525–530 (2015). Article CAS Google Scholar * Whitfield, T. W. et al. Functional analysis of transcription factor binding sites
in human promoters. _Genome Biol._ 13, R50 (2012). Article Google Scholar * Sapkota, Y. et al. A two-stage association study identifies methyl-CpG-binding domain protein 2 gene
polymorphisms as candidates for breast cancer susceptibility. _Eur. J. Hum. Genet._ 20, 682–689 (2012). Article CAS Google Scholar * Sharma, K., Singh, J. & Pillai, P. P. MeCP2
differentially regulate the myelin MBP and PLP protein expression in oligodendrocytes and C6 glioma. _J. Mol. Neurosci._ 65, 343–350 (2018). Article CAS Google Scholar * Ligorio, M. et
al. Stromal microenvironment shapes the intratumoral architecture of pancreatic. _Cancer Cell_ 178, 160–175 (2019). CAS Google Scholar * Colak, S. & Ten Dijke, P. Targeting TGF-β
signaling in cancer. _Trends Cancer_ 3, 56–71 (2018). Article Google Scholar * Zhang, B. et al. Macrophage-expressed CD51 promotes cancer stem cell properties via the TGF-beta1/smad2/3
axis in pancreatic cancer. _Cancer Lett._ 459, 204–215 (2019). Article CAS Google Scholar * Shah, R. R. & Bird, A. P. MECP2 mutations: progress towards understanding and treating Rett
syndrome. _Genome Med._ 9, 17 (2017). Article Google Scholar * He, Y., Tsou, P. S., Khanna, D. & Sawalha, A. H. Methyl-CpG-binding protein 2 mediates antifibrotic effects in
scleroderma fibroblasts. _Ann. Rheum. Dis._ 77, 1208–1218 (2018). PubMed PubMed Central Google Scholar * Henderson, J. et al. Methyl cap binding protein 2: a key epigenetic protein in
systemic sclerosis. _Rheumatology_ 58, 527–535 (2019). Article CAS Google Scholar * Zhang, X. Y., Chang, H. M., Zhu, H., Liu, R. Z. & Leung, P. C. K. BMP6 increases TGF-beta1
production by up-regulating furin expression in human granulosa-lutein cells. _Cell Signal_ 55, 109–118 (2019). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS This study
was supported by grants from the Natural Science Foundation of China [81672402, 81472333, 81301693]; Natural Science Foundation of Jiangsu Province, China [BK20171305]; Research Programs of
Jiangsu Provincial Commission of Health and Family Planning, China [H201434]; the Graduate Research and Innovation Projects of Jiangsu Province [KYCX18_2285]. AUTHOR INFORMATION Author notes
* These authors contributed equally: Huizhi Wang, Jie Li, Junbo He AUTHORS AND AFFILIATIONS * Department of Gastroenterology, Affiliated Hospital of Jiangsu University, Jiangsu University,
438 Jiefang Road, Zhenjiang, 212000, China Huizhi Wang, Jie Li, Junbo He, Yawen Liu, Wen Feng, Hailang Zhou, Meng Zhou, Hong Wei, Ying Lu & Min Xu * Department of Gastroenterology, The
First People’s Hospital of Jingzhou, 8 Aviation Road, Jingzhou, 434000, China Jie Li * Department of Cell Biology, School of Medicine, Jiangsu University, 301 Xuefu Road, Zhenjiang, 212000,
China Wanxin Peng, Fengyi Du & Aihua Gong Authors * Huizhi Wang View author publications You can also search for this author inPubMed Google Scholar * Jie Li View author publications You
can also search for this author inPubMed Google Scholar * Junbo He View author publications You can also search for this author inPubMed Google Scholar * Yawen Liu View author publications
You can also search for this author inPubMed Google Scholar * Wen Feng View author publications You can also search for this author inPubMed Google Scholar * Hailang Zhou View author
publications You can also search for this author inPubMed Google Scholar * Meng Zhou View author publications You can also search for this author inPubMed Google Scholar * Hong Wei View
author publications You can also search for this author inPubMed Google Scholar * Ying Lu View author publications You can also search for this author inPubMed Google Scholar * Wanxin Peng
View author publications You can also search for this author inPubMed Google Scholar * Fengyi Du View author publications You can also search for this author inPubMed Google Scholar * Aihua
Gong View author publications You can also search for this author inPubMed Google Scholar * Min Xu View author publications You can also search for this author inPubMed Google Scholar
CORRESPONDING AUTHORS Correspondence to Aihua Gong or Min Xu. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION FIGURE S1 FIGURE S2 FIGURE
S3 FIGURE S4 FIGURE S5 SUPPLEMENTARY LEGENDS TABLE 1 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Wang, H., Li, J., He, J. _et al._ Methyl-CpG-binding protein 2 drives the Furin/TGF-β1/Smad axis to promote epithelial–mesenchymal transition
in pancreatic cancer cells. _Oncogenesis_ 9, 76 (2020). https://doi.org/10.1038/s41389-020-00258-y Download citation * Received: 19 November 2019 * Revised: 30 June 2020 * Accepted: 15 July
2020 * Published: 26 August 2020 * DOI: https://doi.org/10.1038/s41389-020-00258-y SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative