Therapy-related b-cell acute lymphoblastic leukemia in adults has unique genetic profile with frequent loss of tp53 and inferior outcome
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Access through your institution Buy or subscribe It has been recognized that patients treated with cytotoxic agents for a primary malignancy, autoimmune disorder, or after solid organ
transplant are at increased risk of developing therapy-related hematopoietic neoplasms. These neoplasms include therapy-related myeloid neoplasms (MNs) and less commonly, acute lymphoblastic
leukemia (ALL). While therapy-related MNs have been well characterized and gained recognition as a distinctive high-risk entity by World Health Organization classification of hematopoietic
neoplasms [1], attention has turned to therapy-related ALL relatively recently and more studies are needed to reveal genetic markers of prognostic and therapeutic significance. Diagnostic
criteria for t-ALL cases have not been clearly defined and previous studies often grouped therapy-related B-cell ALL (t-B-ALL) with T-cell ALL and secondary ALL (defined as ALL with a prior
history of other malignancy, regardless of treatment). To the best of our knowledge, only two studies with strict inclusive criteria focused on t-B-ALL [2, 3], with other two [4, 5]
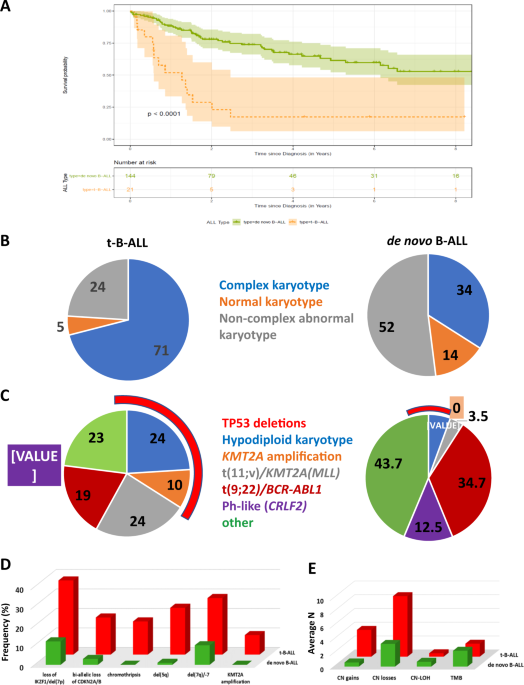
combining B-cell and T-cell ALL cases together. Confirming previously reported findings [2,3,4,5], our cohort of t-B-ALL patients showed significantly higher frequency of high-risk lymphoid
cytogenetic abnormalities, including _KMT2A_ translocations, hypodiploid, or complex karyotype. Although t(9;22)/_BCR-ABL1_ fusion was identified in both cohorts, _CRLF2_ rearrangement,
frequent in de novo _BCR-ABL1_-like B-ALL, was not noted in our t-B-ALL cohort. Some specific genetic markers associated with dismal outcome in B-ALL, i.e., haploinsufficiency of _IKZF1_ and
_TP53_, and biallelic loss of _CDKN2A/CDKN2B_ are highly overrepresented in t-B-ALL compared to de novo B-ALL. Thus, t-B-ALL should be considered as a high-risk lymphoid neoplasm based on
these genetic features. This is a preview of subscription content, access via your institution ACCESS OPTIONS Access through your institution Subscribe to this journal Receive 12 print
issues and online access $259.00 per year only $21.58 per issue Learn more Buy this article * Purchase on SpringerLink * Instant access to full article PDF Buy now Prices may be subject to
local taxes which are calculated during checkout ADDITIONAL ACCESS OPTIONS: * Log in * Learn about institutional subscriptions * Read our FAQs * Contact customer support REFERENCES *
Swerdlow SH, Campo E, Harris NL. World Health Organization classification of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2017. Google Scholar * Saygin C, Kishtagari A,
Cassaday RD, Reizine N, Yurkiewicz I, Liedtke M, et al. Therapy-related acute lymphoblastic leukemia is a distinct entity with adverse genetic features and clinical outcomes. Blood Adv.
2019;3:4228–37. Article CAS Google Scholar * Tang G, Zuo Z, Thomas DA, Lin P, Liu D, Hu Y, et al. Precursor B-acute lymphoblastic leukemia occurring in patients with a history of prior
malignancies: is it therapy-related? Haematologica. 2012;97:919–25. Article Google Scholar * Abdulwahab A, Sykes Jenna, Kamel-Reid S, Chang H, Brandwein JM. Therapy-related acute
lymphoblastic leukemia is more frequent than previously recognized and has a poor prognosis. Cancer. 2012;118:3962–7. Article Google Scholar * Aldoss I, Capelletti M, Park J, Sklavenitis
Pistofidis R, Pillai R, Stiller T, et al. Acute lymphoblastic leukemia as a clonally unrelated second primary malignancy after multiple myeloma. Leukemia. 2019;33:266–70. Article CAS
Google Scholar * McNerney ME, Godley L, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17:5130527. Article Google Scholar *
Andersen MK, Christiansen DH, Kirchhoff M, Pedersen-Bjergaard J. Duplication or amplification of chromosome band 11q23, including the unrearranged MLL gene, is a recurrent abnormality in
therapy-related MDS and AML, and is closely related to mutation of the TP53 gene and to previous therapy with alkylating agents. Genes Chromosom Cancer. 2001;31:33–41. Article CAS Google
Scholar * Fontana MC, Marconi G, Milosevic Feenstra JD, Fonzi E, Papayannidis C, et al. Chromothripsis in acute myeloid leukemia: biological features and impact on survival. Leukemia.
2018;32:1609–20. Article Google Scholar * Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, Layer RM, et al. Breakpoint profiling of 64 cancer genomes reveals numerous complex
rearrangements spawned by homology-independent mechanisms. Genome Res. 2013;23:762–76. Article CAS Google Scholar * Li Y, Schwab C, Ryan S, Papaemmanuil E, Robinson HM, Jacobs P, et al.
Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508:98–102. Article CAS Google Scholar * Forero-Castro M, Robledo C, Benito R,
Abáigar M, África Martín A, Arefi M, et al. Genome-wide DNA copy number analysis of acute lymphoblastic leukemia identifies new genetic markers associated with clinical outcome. PLoS ONE.
2016;11:e0148972. Article Google Scholar * Pang H, Kim Y, Wang H, Lu X, Wang X, Li S. Chromothripsis and chromosomal rearrangements in an adult patient with acute lymphocytic leukemia.
Cancer Genet. 2017;214:P49. Article Google Scholar * Corneaus E, Mullighan C. TP53 mutations in hypodiploid acute lymphoblastic leukemia. Cold Spring Harb Perspect Med. 2017;7:a026286.
Article Google Scholar * Qian M, Cao X, Devidas M, Yang W, Cheng C, Dai Y, et al. TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphobastic leukemia
in children. J Clin Oncol. 2018;36:591–9. Article CAS Google Scholar * Lu C, Xie M, Wendl MC, Wang J, McLellan MD, Leiserson MDM, et al. Patterns and functional implications of rare
germline variants across 12 cancer types. Nat Commun. 2015;6:10086. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS The authors would like to thank the staff of the
cytogenetics and molecular diagnostics laboratories for their technical expertise. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Pathology, Northwestern University Feinberg
School of Medicine, Chicago, IL, USA Liron Barnea Slonim, Juehua Gao, Xinyan Lu, Firas Wehbe, Lawrence Jennings, Yi-Hua Chen & Madina Sukhanova * Department of Hematology and Oncology,
Northwestern University Feinberg School of Medicine, Chicago, IL, USA Madelyn Burkart, Shira N. Dinner, Firas Wehbe & Jessica K. Altman * Department of Pathology and Laboratory Medicine
and Medical Education, Loyola University Chicago Stritch School of Medicine, Chicago, IL, USA Oluwatobi E. Odetola, Firas Wehbe & Kamran M. Mirza * Department of Preventive Medicine
(Health and Biomedical Informatics), Northwestern University Feinberg School of Medicine, Chicago, IL, USA Masha Kocherginsky & Firas Wehbe Authors * Liron Barnea Slonim View author
publications You can also search for this author inPubMed Google Scholar * Juehua Gao View author publications You can also search for this author inPubMed Google Scholar * Madelyn Burkart
View author publications You can also search for this author inPubMed Google Scholar * Oluwatobi E. Odetola View author publications You can also search for this author inPubMed Google
Scholar * Masha Kocherginsky View author publications You can also search for this author inPubMed Google Scholar * Shira N. Dinner View author publications You can also search for this
author inPubMed Google Scholar * Xinyan Lu View author publications You can also search for this author inPubMed Google Scholar * Firas Wehbe View author publications You can also search for
this author inPubMed Google Scholar * Lawrence Jennings View author publications You can also search for this author inPubMed Google Scholar * Jessica K. Altman View author publications You
can also search for this author inPubMed Google Scholar * Kamran M. Mirza View author publications You can also search for this author inPubMed Google Scholar * Yi-Hua Chen View author
publications You can also search for this author inPubMed Google Scholar * Madina Sukhanova View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
MS undertook concept and design; LBS, JG, Y-HC, OEO, KMM, MB, SND, JKA, FW provided study materials; MS and LBS collected and assembled data; MS, LBS, JG, and MK undertook data analysis and
clinical interpretation; MS, LBS, and JG wrote the manuscript; LJ and XL reviewed the manuscript and provided constructive critics, and MS edited the article and provided final approval.
CORRESPONDING AUTHOR Correspondence to Madina Sukhanova. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL MATERIALS
RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Barnea Slonim, L., Gao, J., Burkart, M. _et al._ Therapy-related B-cell acute lymphoblastic leukemia in
adults has unique genetic profile with frequent loss of _TP53_ and inferior outcome. _Leukemia_ 35, 2097–2101 (2021). https://doi.org/10.1038/s41375-020-01061-9 Download citation * Received:
21 July 2020 * Revised: 01 September 2020 * Accepted: 05 October 2020 * Published: 21 October 2020 * Issue Date: July 2021 * DOI: https://doi.org/10.1038/s41375-020-01061-9 SHARE THIS
ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard
Provided by the Springer Nature SharedIt content-sharing initiative