Prmt7 regulates the JAK/STAT/Socs3 signaling pathway in postmenopausal cardiomyopathy
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Protein arginine methyltransferases (PRMTs) modulate diverse cellular processes, including stress responses. The present study explored the role of Prmt7 in protecting against
menopause-associated cardiomyopathy. Mice with cardiac-specific Prmt7 ablation (cKO) exhibited sex-specific cardiomyopathy. Male cKO mice exhibited impaired cardiac function, myocardial
hypertrophy, and interstitial fibrosis associated with increased oxidative stress. Interestingly, female cKO mice predominantly exhibited comparable phenotypes only after menopause or
ovariectomy (OVX). Prmt7 inhibition in cardiomyocytes exacerbated doxorubicin (DOX)-induced oxidative stress and DNA double-strand breaks, along with apoptosis-related protein expression.
Treatment with 17β-estradiol (E2) attenuated the DOX-induced decrease in Prmt7 expression in cardiomyocytes, and Prmt7 depletion abrogated the protective effect of E2 against DOX-induced
cardiotoxicity. Transcriptome analysis of ovariectomized wild-type (WT) or cKO hearts and mechanical analysis of Prmt7-deficient cardiomyocytes demonstrated that Prmt7 is required for the
control of the JAK/STAT signaling pathway by regulating the expression of suppressor of cytokine signaling 3 (Socs3), which is a negative feedback inhibitor of the JAK/STAT signaling
pathway. These data indicate that Prmt7 has a sex-specific cardioprotective effect by regulating the JAK/STAT signaling pathway and, ultimately, may be a potential therapeutic tool for heart
failure treatment depending on sex.
Diverse pathological conditions, such as hypertension, myocardial infarction, myocarditis, or heart valve disease, cause cardiac remodeling, leading to cardiomyocyte hypertrophy, fibrosis,
necrosis, and eventually heart failure1,2. Interestingly, men are more susceptible to heart disease than women are; however, postmenopausal women are at greater risk than premenopausal women
are for developing cardiovascular diseases, even though there is still a lower incidence of heart disease in postmenopausal women than in similarly aged men3,4,5.
Based on the discrepancy in the causes of cardiovascular disease according to sex and reproductive age, the sex hormone E2 is believed to be associated with protection against heart
disease5,6. To date, multiple studies have been conducted to investigate the protective mechanism of E2 against cardiovascular diseases6. E2 enhances mitochondrial structure and function by
activating the PI3K/ERK signaling pathway and reduces oxidative stress by promoting the generation of the antioxidants SOD2 and hydrogen sulfide7. Additionally, E2 is known to be involved in
extracellular matrix (ECM) remodeling by modulating fibroblasts via estrogen receptors and the MAPK signaling pathway8. Additionally, E2 inhibits fibroblast proliferation and the expression
of profibrotic genes, such as Col1A1 (encoding Collagen I), Col3A1 (encoding Collagen III), and Fbrs (encoding Fibrosin I), resulting in the suppression of fibrotic deposition, which is
closely connected to hindering the relaxation and contraction of the heart9.
Protein arginine methylation is an important type of post-translational modification mediated by PRMTs and is commonly associated with normal cellular processes, including DNA repair, cell
cycle regulation, transcription, mRNA splicing, and signal transduction10,11. PRMTs can be classified into three types according to their catalytic activity (I-III). The type I enzymes
PRMT1, PRMT2, PRMT3, CARM1 (also known as PRMT4), PRMT6 and PRMT8 dimethylate their substrates asymmetrically. The type II enzymes PRMT5 and PRMT9 are symmetrical dimethyltransferases. The
only member of the type III group is PRMT7, which can solely monomethylate arginine. Among them, Prmt7 has a unique structure consisting of two tandem PRMT modules, and there is growing
information on the biological functions of Prmt7 in terms of gene expression, differentiation, senescence, and stress responses12,13.
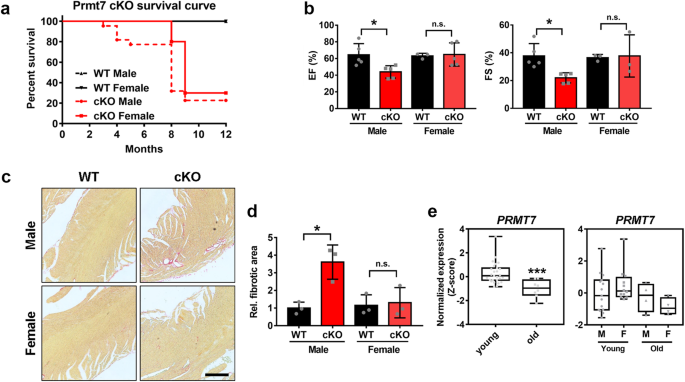
In our previous study, we demonstrated that Prmt7 plays a protective role in cardiomyocytes against cardiac remodeling in heart failure14. Interestingly, during this study, cardiomyopathy
resulting from Prmt7 deficiency was found only in young male mice and not in young female mice. This sex difference in the cardiomyopathy phenotype prompted us to question the distinct role
of Prmt7 in female-related cardiac function. In this study, we found that cardiac-specific Prmt7 deficiency in postmenopausal or ovariectomized mice was associated with oxidative stress,
apoptosis, and fibrosis, ultimately leading to severe cardiomyopathy. Additionally, there was a correlation between Prmt7 and E2, and Prmt7 was found to be required for the complete
activation of E2 in cardiomyocytes. Analysis of the transcriptome and underlying mechanism demonstrated that Prmt7 plays a role in regulating the expression of Socs3, which functions as a
negative feedback inhibitor in the JAK/STAT signaling pathway.
Taken together, our data have implications for understanding the cardioprotective role of Prmt7 in postmenopause-related cardiomyopathy and ultimately allow for the identification of new
prognostic indicators or novel therapeutic strategies for the treatment of heart failure.
Prmt7+/- mice were maintained as previously described15. Briefly, C57BL/6N-Tyrc-Brd Prmt7tm1a (EUCOMM) wtsi/WtsiCnbc mice purchased from the Sanger Institute were backcrossed onto a C57BL/6
J background for at least 10 generations. Littermate wild-type mice were used as control groups for Prmt7-/- mice in all the experiments. For generation of cardiac-specific Prmt7 null mice,
Prmt7Tm1c/Tm1c (Prmt7f/f) mice were crossbred with mice harboring the Myh6-Cre transgene [Tg(Myh6-cre)2182Mds/J] (Jackson Laboratory, Bar Harbor, ME). Genotyping was performed as previously
described15. To assess the effect of Prmt7 on cardiomyopathy after menopause, we utilized 10-month-old female littermates obtained through heterozygous breeding. For the development of OVX,
the ovaries of 2-month-old female mice were surgically excised, and 7 days later, 25 mg/kg DOX (Tocris Bioscience, Bristol, UK) was administered to induce cardiotoxicity. The mice were
subjected to echocardiography 4 days after administration and were sacrificed the next day.
For echocardiographic analysis, the mice were anesthetized with 1–2% (vol/vol) isoflurane. Echocardiography was performed one day before the mice were sacrificed using a Vevo LAZR-X
photoacoustic imaging system (Fujifilm VisualSonics, Toronto, Canada). Heart rate was monitored and generally maintained at 400-500 beats per minute. Analyses of M-mode images derived from
the short-axis view of the left ventricle (LV) were performed to calculate the ejection fraction (EF) and fractional shortening (FS).
For animal studies, drug administration was approved by the Institutional Animal Care and Use Committee (IACUC) of the Sungkyunkwan University School of Medicine (SUSM) and carried out
according to ethical guidelines.
H9c2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Billings, MT) supplemented with 10% fetal bovine serum (FBS; Gibco, Billings, MT) and 1% penicillin/streptomycin
(Thermo Fisher Scientific, Waltham, MA). Newborn rat ventricular myocytes (NRVMs) were isolated from neonatal Sprague–Dawley (SD, 1–2 days) rat heart tissues as previously described14. For
analysis of the correlation between Prmt7 and E2 under cardiotoxic conditions, H9c2 cells were exposed to 10 nM E2 (Sigma‒Aldrich) and/or 1 μM DOX for 16 h. Transfection studies were
performed as previously described14. Polyethylenimine (1 mg/ml; Sigma‒Aldrich, St. Louis, MO) was used to transfect pcDNA-PRMT7 or pcDNA-HA-PRMT7 into H9c2 cells. These overexpression
plasmids encode human PRMT715. For Prmt7 depletion, a lentiviral vector containing either shScrambled or shPrmt7 was utilized, as previously described16. For analysis of the effects of Prmt7
inhibition, cells were treated with 50 μM DS-437 (Sigma‒Aldrich), a dual inhibitor of Prmt5 and Prmt7, or 1 μM SGC-8158 (Sigma‒Aldrich) for 24 h. For activation of the JAK/STAT signaling
pathway, H9c2 cells were treated with 104 units/ml of LIF (Sigma‒Aldrich) for 30 min.
Immunoblotting and immunostaining were performed as previously described14. Briefly, cultured cells were lysed using lysis buffer (50 mM Tris-HCl (pH 7.4), 1.5 mM MgCl2, 150 mM NaCl, 1 mM
EGTA, 1% Triton X-100, and complete protease inhibitor cocktail) and analyzed by standard western blotting. The antibodies used in this study are as follows: Prmt7 (Abcam, ab179822, 1:500
dilution), cleaved Caspase-3 (Cell Signaling, Cat# 9664, 1:500 dilution), γH2AX (Cell Signaling, Cat# 9718, 1:500 dilution), HA (Cell Signaling, Cat# 3724, 1:1000 dilution), β-actin (Cell
Signaling, #4970, 1:500 dilution), pSTAT3 (Cell Signaling, Cat# 9145, 1:500 dilution), STAT3 (Cell Signaling, Cat# 9139, 1:500 dilution), and HSP90 (Santa Cruz, sc-7947, 1:1000 dilution).
The quantification of protein levels was performed by signal density analysis using the ImageJ program, after which the protein levels were normalized to the level of a loading control, such
as β-actin or HSP90.
Quantitative RT‒PCR was performed as previously described14. Total RNA from mouse hearts was extracted with easy-BLUE reagent (iNtRON, Seongnam, Republic of Korea) following the
manufacturers’ instructions. cDNAs were synthesized from 0.5 μg of total RNA by using a PrimeScript RT reagent kit (TaKaRa, Shiga, Japan) according to the manufacturer’s instructions. Gene
expression was quantified by using SYBR Premix Ex Taq (TaKaRa) on a Thermal Cycler Dice Real-Time System (TaKaRa, TP800) following the manufacturer’s instructions. The sequences of primers
used in this study are indicated below; IL-1α (Forward) 5’-GCCCGTGTTGCTGAAGGAGT-3’ (Reverse) 5′-CATAGAGGGCAGTCCCCGTG-3′; IL-6 (Forward) 5′-TACCACTTCACAAGTCGGAGGC-3′ (Reverse)
5′-CTGCAAGTGCATCATCGTTGTTC-3′; IL-18 (Forward) 5′-GACAGCCTGTGTTCGAGGATATG-3′ (Reverse) 5′-TGTTCTTACAGGAGAGGGTAGAC-3′; IFN-γ (Forward) 5′-CAGCAACAGCAAGGCGAAAAAGG-3′ (Reverse)
5′-TTTCCGCTTCCTGAGGCTGGAT-3′; TNF-α (Forward) 5′-AAATGGGCTCCCTCTCATCAGTTC-3′ (Reverse) 5′- TCTGCTTGGTGGTTTGCTACGAC-3′; Socs3 (Forward) 5′-GACCTGTCTACAGCTCTCCGTC-3′ (Reverse)
5′-CTGCGCCTCCTATGGTCCC-3′; 18S rRNA (Forward) 5′-GTAACCCGTTGAACCCCATT-3′ (Reverse) 5′-CCATCCAATCGGTAGTAGCG-3′; and L-32 (Forward) 5′-GGCCTCTGGTGAAGCCCAAGATCG-3′ (Reverse)
5′-CCTCTGGGTTTCCGCCAGTTTCGC-3′.
The transcriptome of PRMT7-expressing heart tissue from young and old human males and females was analyzed with the GSE36961 dataset from the Gene Expression Omnibus (GEO;
http://www.ncbi.nlm.nih.gov/geo).
High-throughput sequencing was performed as single-end 75 sequencing using an Illumina NExtSeq 500 (Ebiogen, Seoul, Korea). The analysis of RNA sequencing data was performed by using ExDEGA
v3.0 (Ebiogen) and Morpheus (http://software.broadinstitute.org/morpheus/). Global gene expression was assessed via Reactome with gene set enrichment analysis (GSEA)
(http://www.gsea-msigdb.orb/gsea/msigdb/index.jsp) using the MsigDB database v7.2 (>1.3-fold, RC log2 > 1.0, P