C-terminal truncated dystrophin identified in skeletal muscle of an asymptomatic boy with a novel nonsense mutation of the dystrophin gene
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Mutations that cause premature stop codons in the dystrophin gene lead to a complete loss of dystrophin from skeletal muscle, resulting in severe Duchenne muscular dystrophy. Here,
a C-terminally truncated dystrophin resulting from a novel nonsense mutation is shown for the first time to be localized to the muscle plasma membrane. An asymptomatic 8-y-old boy was
examined for dystrophin in skeletal muscle because of high serum creatine kinase activity. Remarkably, no dystrophin labeling was seen with an MAb against the C-terminal domain, suggesting
the presence of an early stop codon in the dystrophin gene. Labeling with an antibody specific to the N-terminal domain, however, revealed weak, patchy, and discontinuous staining,
suggesting limited production of a truncated form of the protein. Molecular analysis revealed a novel nonsense mutation (Q3625X) as a result of a single nucleotide change in the
patient's genomic DNA (C10873T), leaving 1.6% of dystrophin gene product unsynthesized at the C terminus. Dystrophin mRNA analysis did not show rescue of the nonsense mutation as a
result of exon-skipping by an alternative splicing mechanism. This is the first report of an asymptomatic dystrophinopathy with a nonsense mutation in the dystrophin gene. SIMILAR CONTENT
BEING VIEWED BY OTHERS NATIVE DGC STRUCTURE RATIONALIZES MUSCULAR DYSTROPHY-CAUSING MUTATIONS Article 11 December 2024 ETIOLOGY OF GENETIC MUSCLE DISORDERS INDUCED BY MUTATIONS IN FAST AND
SLOW SKELETAL MYBP-C PARALOGS Article Open access 01 March 2023 _DAG1_ HAPLOINSUFFICIENCY IS ASSOCIATED WITH SPORADIC AND FAMILIAL ISOLATED OR PAUCI-SYMPTOMATIC HYPERCKEMIA Article 04
January 2024 MAIN The severe Duchenne muscular dystrophy (DMD) and the more benign Becker muscular dystrophy (BMD) are allelic conditions characterized by progressive muscular degeneration
and wasting accompanied by an elevation of serum creatine kinase (CK). DMD is a rapidly progressive disease, with those affected starting to show muscle weakness at 4–5 y of age and losing
the ability to walk independently before the age of 12 y. BMD has a slower rate of progression; affected individuals remain ambulatory beyond the age of 16 y, and a few may lead near-normal
lives (1). DMD and BMD are caused by mutation of the dystrophin gene, which encodes a 14-kb mRNA that consists of 79 exons. The gene is the largest in humans and covers >3000 kb on the X
chromosome (2,3). DMD and BMD are the most common genetic muscle diseases, affecting >1 in 3,500 male births. Two thirds of DMD/BMD patients have deletion or duplication mutations of the
dystrophin gene, and their clinical progression can be predicted by whether the deletion or duplication maintains (in-frame) or disrupts (out-of-frame) the translational reading frame (the
reading-frame rule) (4). Dystrophin is absent from skeletal muscle of DMD, because the dystrophin that is produced is truncated as a result of the premature stop codon and therefore is
unstable, whereas in BMD, dystrophin that contains internal in-frame deletions produces protein that can be detected (5). Single-base nonsense mutations have been suspected in DMD patients
who do not show deletion/duplication mutations. However, detection of such defects in individual DMD patients is very difficult as a result of the large size of the gene. More than 100
nonsense mutations have been reported at various points over a 14-kb length of the dystrophin mRNA (http://www.dmd.nl). Despite this wide variation in coding potential (0–98.6% of the
full-length protein), these truncating mutations are associated with a surprisingly uniform severity of the DMD phenotype (6). However, a limited number of single-base nonsense mutations
have been reported in patients with mild BMD that showed skipping of the exon encoding the mutation, thus producing an in-frame mRNA (7–11). Dystrophin is a cytoskeletal protein that is
implicated in membrane stability and in communication between the extracellular matrix and the inner cytoskeleton (12,13). The protein, which consists of 3685 amino acids, is divided into
four distinct domains: an N-terminal domain, a large rod-like domain of 24 spectrin-like repeats that occupies >70% of its length, a cysteine-rich domain, and, finally, a C-terminal
domain (2,14). Studies conducted on DMD/BMD patients suggest that the N-terminal, cysteine-rich, and C-terminal domains are essential for dystrophin's function (15,16). Notably, the
C-terminal domain, which consists of 416 amino acids encoded by 13 exons, shows sequence similarity with only two other dystrophin-related proteins and is considered to exert
dystrophin's specific function (14,17,18). In fact, in-frame deletions that extend into the C-terminal domain have been reported to result in DMD, whereas large in-frame deletions of
the rod domain result in BMD (16). Here we report a C-terminally truncated dystrophin caused by a mutation in an asymptomatic boy with high CK activity. We propose that nonsense mutations of
the dystrophin gene can result in a wide variety of clinical phenotypes. METHODS CASE. The proband (KUDN 02765682) was an 8-y-old boy. His family history disclosed no neuromuscular disease.
He started to walk independently at 1 y of age, and his motor development was normal. He had a history of transient muscle weakness. At the age of 3 y, he complained of pain in the lower
legs without any predisposing signs or symptoms and lost the ability to stand up and walk by himself. His serum CK was found to be 4901 IU/L (normal <169 IU/L). The muscle weakness
persisted for 1 wk but disappeared spontaneously and completely. During the following period, his serum CK remained elevated but showed a strong fluctuation in value, ranging from 1,607 to
21,100 IU/L. Despite his high CK, he did not show any muscle weakness. At the age of 5 y, he was referred to Kobe University Hospital for examination of his elevated serum CK activity. His
mental development was normal. On physical examination, there was no Gower's sign, walking abnormality, or pseudohypertrophy of the legs. An electromyogram disclosed myogenic changes. A
chest x-ray, electrocardiography, and echocardiography failed to reveal cardiac abnormalities. To clarify the cause of the elevation in serum CK and myogenic pattern in electromyogram, a
quadriceps muscle biopsy was carried out after obtaining informed consent. The protocols of this study were approved by our ethical committee. IMMUNOHISTOCHEMICAL ANALYSIS. The muscle biopsy
sample was examined pathologically and immunohistochemically. An indirect immunofluorescence analysis was performed using three dystrophin antibodies that recognize the N-terminal
(NCL-Dys3), the rod (NCL-Dys1), and the C-terminal (NCL-Dys2) domains of dystrophin (Novocastra Laboratories, Newcastle upon Tyne, UK) (5,19). Furthermore, utrophin, β-dystroglycan,
γ-sarcoglycan (Novocastra Laboratories Ltd, Newcastle upon Tyne, UK), laminin α2 (Chemicon International Inc., Temecula, CA), and α-dystroglycan (Upstate Biotechnology, Lake Placid, NY) were
also stained using their respective antibodies. Control skeletal muscle tissue was obtained with informed consent and was simultaneously stained with the same panel of antibodies. Western
blot analysis of dystrophin using an MAb that recognizes the C-terminal domain was performed by Athena Diagnostics (Worcester, MA). ANALYSIS OF THE DYSTROPHIN GENE. For mutational analysis
of the dystrophin gene, blood samples were obtained from the index case and family members after obtaining informed consent. DNA was isolated by standard phenol-chloroform extraction
methods. For screening for deletion mutations, 19 deletion-prone exons were amplified from the genomic DNA by PCR essentially according to methods described previously (20). Southern blot
analysis using dystrophin cDNA as a probe was performed with _Hin_dIII restriction enzyme–digested DNA as a template, as described by Koenig _et al._ (21). For analyzing genomic mutations,
the region that encompasses exon 76 was amplified by PCR with g76F:5′-GGAGGGCTTCTAAAGTAGG-3′ as the forward primer and g76r:5′-ATGTCCCTGTAATACGACTCTACC-3′ as the reverse primer under
conditions described elsewhere (20). ANALYSIS OF DYSTROPHIN MRNA. Reverse-transcription PCR (RT-PCR) was used to analyze the dystrophin mRNA expressed in lymphocytes or skeletal muscle as
described by Roberts _et al._ (22,23). Full-length dystrophin cDNA was amplified as 10 separate, partially overlapping fragments and sequenced directly. For obtaining a fragment showing
aberrant splicing, including exon 76 skipping, a region that encompasses exons 70–79 was amplified using a forward primer corresponding to a segment of exon 70
(70f:5′-CAGGAGAAGATGTTCGAGAC-3′) and a reverse primer complementary to a segment of exon 79 (5f:5′-ATCATCTGCCATGTGGAAAAG-3′). SEQUENCING OF THE AMPLIFIED PRODUCT. The amplified product was
purified and subjected to sequencing either directly or after subcloning into a pT7 blue T vector (Novagen, Madison, WI) (24). The DNA sequence was determined using an automated DNA
sequencer (model 373A; Applied Biosystems, Foster City, CA). RESULTS For elucidating the cause of the elevation in serum CK, the biopsied muscle sample was examined pathologically.
Microscopic examination disclosed slight dystrophic changes such as size variation in muscle fibers, fibers with central nuclei, and degenerated and regenerated fibers. Immunofluorescence
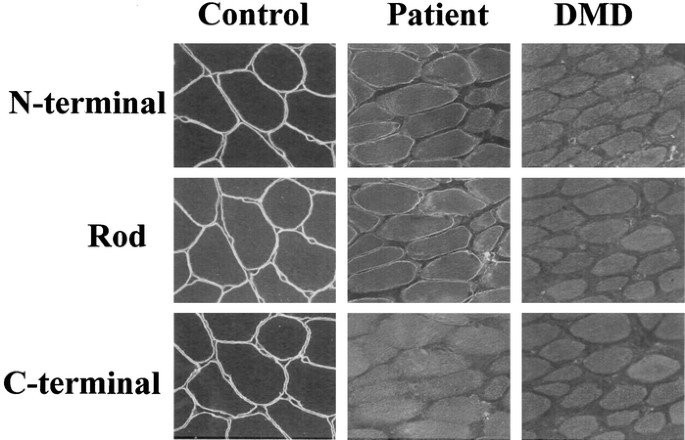
staining for dystrophin revealed a complete absence of C-terminal domain labeling (Fig. 1). In contrast, both N-terminal and rod-domain staining was weak, patchy, and discontinuous along the
plasma membrane (Fig. 1). These findings clearly indicated dystrophinopathy, but the patterns of dystrophin staining were not typical for either DMD or BMD. Western blot analysis of
dystrophin using an antibody that recognizes the C-terminal domain of dystrophin revealed no significant bands (data not shown). These staining patterns indicate that a nonsense mutation in
dystrophin is present in this patient, leading to production of a protein truncated somewhere upstream of the C-terminal epitope recognized by the aforementioned antibody. For clarifying the
molecular pathogenesis of the abnormal dystrophin, the dystrophin gene was scanned for mutations. Neither PCR amplification of 19 selected exons nor Southern blot analysis of the dystrophin
gene revealed any gross gene rearrangements. The possibility of deletion mutation therefore seemed unlikely. To find a single-base mutation, we analyzed dystrophin mRNA extracted from
peripheral lymphocytes using RT-PCR as described previously (22). Ten fragments covering the full-length dystrophin cDNA could be amplified as normal-sized products. Direct sequencing of a
fragment that encompasses exons 70–79 disclosed a single nucleotide change: a transition from a cytosine to a thymine at nucleotide 10873 (C10873T) in exon 76 (14). The same nucleotide
change (C10873T) was present not only in his muscle dystrophin mRNA (Fig. 2) but also in his genomic DNA (data not shown) (25). His mother was found to be a carrier of the same mutation
(data not shown). Because sequencing of other fragments of dystrophin cDNA disclosed no other significant nucleotide changes, it was concluded that this mutation (C10873T) is the cause of
the dystrophinopathy. The nucleotide change converted a CAG codon, which encodes glutamine at the 3625th amino acid position, to a stop TAG codon (Q3625X; (Fig. 2). Therefore, a truncated
dystrophin lacking 60 amino acids at its C terminus (1.6% of the total dystrophin sequence) was expected to be produced. This truncation of dystrophin is compatible with the failure of the
C-terminus–specific antibody to label the protein (Fig. 1), because this antibody recognizes amino acids 3669–3685, an epitope that is downstream of the premature stop codon (Q3625X).
However, that dystrophin did stain positively with antibodies against its N-terminal and rod domains (Fig. 1) does not seem consistent with this truncation mutant, because other truncated
dystrophin mutants have been found to be very unstable and undetectable immunohistochemically (4,6). In other cases, it has been hypothesized that the positive staining of dystrophin is the
result of rescue of nonsense mutations by exon skipping or aberrant splicing (8,26). However, the RT-PCR–amplified product encompassing exons 70–79 disclosed only one visible band upon
agarose gel electrophoresis (Fig. 2). In addition, both direct sequencing and sequencing after subcloning the product confirmed the presence of normal exon structure, indicating that only
one mRNA was produced from the mutated gene. These observations do not support the possibility of exon-76 skipping or aberrant splicing. Although the patient in our case harbored a Q3625X
nonsense mutation, his clinical phenotype was unusually mild. Utrophin, a dystrophin-related protein, has been proposed to compensate for the function of dystrophin (27). Therefore,
overexpression of utrophin might account for the clinical phenotype seen in the present case. The expression of utrophin was studied in muscle (Fig. 3) and was not found to be elevated in
the index case in comparison with that typically seen in DMD. Therefore, enhanced expression of utrophin does not seem, to be modifying the clinical phenotype. Furthermore, the
dystrophin-dystroglycan axis was examined (Fig. 3). Laminin α2, an extracellular matrix protein, stained weakly. Neither α-dystroglycan, an extracellular β-dystroglycan–binding protein, nor
β-dystroglycan, a transmembrane dystrophin-binding protein, was stained. γ-Sarcoglycan, a member of the sarcoglycan complex, was stained very weakly. All of these staining patterns were
similar to those found in DMD (Fig. 3), indicating no difference in the stabilization of the dystrophin-dystroglycan axis from that observed in DMD (Fig. 3). Therefore, no explanation for
the mild phenotype was obtained through studies of protein staining. DISCUSSION A novel nonsense mutation (Q3625X) in the dystrophin gene was identified in a Japanese boy who was as yet
asymptomatic at the age of 8 y. Although a severe DMD phenotype would be expected to develop from his mutation type, his clinical course has been extraordinarily mild. The case has raised an
important question to be answered: What is the mechanism that determines the severity of the dystrophic phenotype? A somatic mosaic for a nonsense mutation has been shown to attenuate the
clinical phenotype (28). However, this possibility seems to be excluded in the index case for the following reasons: _1_) the mutation was inherited through the mother, and _2_) a single
genomic clone harboring C10873T was obtained not only from his lymphocytes but also from his muscle (data not shown). To rule out this possibility unequivocally, it is necessary to examine
other muscle tissues, but this has not yet been done. Another possible attenuating mechanism would be the modification of mRNA by either exon skipping or aberrant splicing, which would
remove the nonsense mutation and produce a more complete dystrophin mRNA. Examples of exon skipping have been reported in nonsense mutations identified in exons 25, 27, 29, and 72 of the
dystrophin gene (7–11), and aberrant splicing has been reported in intermediate dystrophinopathy (26). In our case, however, not only the RT-PCR product of dystrophin mRNA but also
subcloning sequencing disclosed the existence of only one kind of mRNA consisting of a normal exon structure (Fig. 2). The possibility of either exon skipping or aberrant splicing thus was
ruled out. Therefore, the discrepancy between genotype and phenotype could not be explained at the mRNA level. Elevated expression of utrophin has been speculated to convert a severe
phenotype to a mild one without affecting dystrophin expression (27). However, level of utrophin expression was the same in our case as in DMD (Fig. 3). Furthermore, the staining of proteins
encompassing the dystrophin-dystroglycan axis was identical to that seen in DMD (Fig. 3). These similarities show that protein-level changes do not underlie the observed phenotypic
differences. The truncated dystrophin produced in the index case seems to be unusually stable, as demonstrated by the weak but significant staining of the N-terminal and rod domains (Fig.
1). This may be because the truncated dystrophin retains functionally important binding sites, such as actin binding sites in the N-terminal and rod domains (29,30), a β-dystroglycan binding
site in the cysteine-rich domain (31), and syntrophin and dystrobrevin binding sites and a phosphorylation site in the C-terminal domain (Fig. 4) (32–35). In fact, it has been demonstrated
that dystrophin lacking the amino acids encoded by exons 71–78 is stable in muscle membranes of the _mdx_ mouse, an animal model of DMD (36). However, this hypothesis is not supported by a
previous report that Q3625X, a nonsense mutation just 10 amino acids downstream of the one reported here (Q3635X), gave rise to clinically typical DMD (6) (Fig. 4). Furthermore, examination
of laminin alpha 2, α- and β-dystroglycans, and γ-sarcoglycan disclosed no difference in their staining patterns between our case and DMD (Fig. 3), indicating that augmented stabilization of
these proteins does not contribute to the mildness of the phenotype. Clearly, further study is required to clarify these complex results. Activation of transcription of the dystrophin gene
may lead to overproduction of dystrophin mRNA. It has been proposed that an abnormality in a transcription factor(s) or in its binding site in the promoter of the dystrophin gene is a factor
in phenotypic severity. In fact, mutation of the MYF6 gene results in a severe phenotype of BMD (37). However, modifier(s) that make the phenotype mild have not been reported to date
(38,39), although _mdx_ mice characterized by dystrophin deficiency do not show a severe DMD phenotype (40). We are now following up the index case, and a future study analyzing not only the
dystrophin gene but also other genes may clarify the molecular mechanism explaining his mild phenotype. ABBREVIATIONS * BMD: Becker muscular dystrophy * CK: creatine kinase * DMD: Duchenne
muscular dystrophy REFERENCES * Emery AEH 1993 Duchenne Muscular Dystrophy. Oxford University Press, Oxford, pp 26–44 * Ahn AH, Kunkel LM 1993 The structural and functional diversity of
dystrophin. _Nat Genet_ 3: 283–291 Article CAS Google Scholar * Nishio H, Takeshima Y, Narita N, Yanagawa H, Suzuki Y, Ishikawa Y, Minami R, Nakamura H, Matsuo M 1994 Identification of a
novel first exon in the human dystrophin gene and of a new promoter located more than 500 kb upstream of the nearest known promoter. _J Clin Invest_ 94: 1037–1042 Article CAS Google
Scholar * Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM 1988 An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus.
_Genomics_ 2: 90–95 Article CAS Google Scholar * Arahata K, Ishiura S, Ishiguro T, Tsukahara T, Suhara Y, Eguchi C, Ishihara T, Nonaka I, Ozawa E, Sugita H 1988 Immunostaining of skeletal
and cardiac muscle surface membrane with antibody against Duchenne muscular dystrophy peptide. _Nature_ 333: 861–863 Article CAS Google Scholar * Prior TW, Bartolo C, Pearl KP, Papp AC,
Snyder PJ, Sedra MS, Burghes AHM, Mendell JR 1995 Spectrum of small mutations in the dystrophin coding region. _Am J Hum Genet_ 57: 22–33 Article CAS Google Scholar * Barbieri AM, Soriani
N, Ferlini A, Michelato A, Ferrari M, Carrera P 1996 Seven novel additional small mutations and a new alternative splicing in the human dystrophin gene detected by heteroduplex analysis and
restricted RT-PCR heteroduplex analysis of illegitimate transcripts. _Eur J Hum Genet_ 4: 183–187 Article CAS Google Scholar * Shiga N, Takeshima Y, Sakamoto H, Inoue K, Yokota Y,
Yokoyama M, Matsuo M 1997 Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible
for Becker muscular dystrophy. _J Clin Invest_ 100: 2204–2210 Article CAS Google Scholar * Melis MA, Muntoni F, Cau M, Loi D, Puddu A, Boccone L, Mateddu A, Cianchetti C, Cao A 1998 Novel
nonsense mutation (C–>A nt 10512) in exon 72 of dystrophin gene leading to exon skipping in a patient with a mild dystrophinopathy. _Hum Mutat_ 1( suppl): S137–S138 Article Google
Scholar * Ginjaar IB, Kneppers AL, v d Meulen JD, Anderson LV, Bremmer-Bout M, van Deutekom JC, Weegenaar J, den Dunnen JT, Bakker E 2000 Dystrophin nonsense mutation induces different
levels of exon 29 skipping and leads to variable phenotypes within one BMD family. _Eur J Hum Genet_ 8: 793–796 Article CAS Google Scholar * Fajkusova L, Lukas Z, Tvrdikova M, Kuhrova VV,
Hajek J, Fajkus J 2001 Novel dystrophin mutations revealed by analysis of dystrophin mRNA: alternative splicing suppresses the phenotypic effect of a nonsense mutation. _Neuromuscul Disord_
11: 133–138 Article CAS Google Scholar * O'Brien KF, Kunkel LM 2001 Dystrophin and muscular dystrophy: past, present, and future. _Mol Genet Metab_ 74: 75–88 Article CAS Google
Scholar * Burton EA, Davies KE 2002 Muscular dystrophy—reason for optimism?. _Cell_ 108: 5–8 Article CAS Google Scholar * Koenig M, Monaco AP, Kunkel LM 1988 The complete sequence of
dystrophin predicts a rod-shaped cytoskeletal protein. _Cell_ 53: 219–228 Article CAS Google Scholar * Passos-Bueno MR, Vainzof M, Marie SK, Zatz M 1994 Half the dystrophin gene is
apparently enough for a mild clinical course: confirmation of its potential use for gene therapy. _Hum Mol Genet_ 3: 919–922 Article CAS Google Scholar * Takeshima Y, Nishio H, Narita N,
Wada H, Ishikawa Y, Ishikawa Y, Minami R, Nakamura H, Matsuo M 1994 Amino-terminal deletion of 53% of dystrophin results in an intermediate Duchenne-Becker muscular dystrophy phenotype.
_Neurology_ 44: 1648–1651 Article CAS Google Scholar * Matsumura K, Ervasti J, Ohlendieck K, Kahl S, Campbell K 1992 Association of dystrophin-related protein with dystrophin-associated
proteins in mdx mouse muscle. _Nature_ 360: 588–591 Article CAS Google Scholar * Hugnot JP, Gilgenkrantz H, Vincent N, Chafey P, Morris GE, Monaco AP, Berwald-Netter Y, Koulakoff A,
Kaplan JC, Kahn A, Chelly J 1992 Distal transcript of the dystrophin gene initiated from an alternative first exon and encoding a 75-kDa protein widely distributed in nonmuscle tissues.
_Proc Natl Acad Sci USA_ 89: 7506–7510 Article CAS Google Scholar * Yagi M, Takeshima Y, Wada H, Nakamura H, Matsuo M 2003 Two alternative exons can result from activation of the cryptic
splice acceptor site deep within intron 2 of the dystrophin gene in a patient with as yet asymptomatic dystrophinopathy. _Hum Genet_ 112: 164–170 CAS PubMed Google Scholar * Matsuo M,
Masumura T, Nakajima T, Kitoh Y, Takumi T, Nishio H, Koga J, Nakamura H 1990 A very small frame-shifting deletion within exon 19 of the Duchenne muscular dystrophy gene. _Biochem Biophys Res
Commun_ 170: 963–967 Article CAS Google Scholar * Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM 1987 Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA
and preliminary genomic organization of the DMD gene in normal and affected individuals. _Cell_ 50: 509–517 Article CAS Google Scholar * Roberts RG, Barby TF, Manners E, Bobrow M, Bentley
DR 1991 Direct detection of dystrophin gene rearrangements by analysis of dystrophin mRNA in peripheral blood lymphocytes. _Am J Hum Genet_ 49: 298–310 CAS PubMed PubMed Central Google
Scholar * Matsuo M, Nishio H, Kitoh Y, Francke U, Nakamura H 1992 Partial deletion of a dystrophin gene leads to exon skipping and to loss of an intra-exon hairpin structure from the
predicted mRNA precursor. _Biochem Biophys Res Commun_ 182: 495–500 Article CAS Google Scholar * Surono A, Takeshima Y, Wibawa T, Ikezawa M, Nonaka I, Matsuo M 1999 Circular dystrophin
RNAs consisting of exons that were skipped by alternative splicing. _Hum Mol Genet_ 8: 493–500 Article CAS Google Scholar * Ito T, Takeshima Y, Yagi M, Kamei S, Wada H, Matsuo M 2003
Analysis of dystrophin mRNA from skeletal muscle but not from lymphocytes led to identification of a novel nonsense mutation in a carrier of Duchenne muscular dystrophy. _J Neurol_ 250:
581–587 Article CAS Google Scholar * Adachi K, Takeshima Y, Wada H, Yagi M, Nakamura H, Matsuo M 2003 Heterogous dystrophin mRNAs produced by a novel splice acceptor site mutation in
intermediate dystrophinopathy. _Pediatr Res_ 53: 125–131 Article CAS Google Scholar * Blake DJ, Weir A, Newey SE, Davies KE 2002 Function and genetics of dystrophin and dystrophin-related
proteins in muscle. _Physiol Rev_ 82: 291–329 Article CAS Google Scholar * Prior TW, Bartolo C, Papp AC, Snyder PJ, Sedra MS, Burghes AH, Mendell JR 1996 Nonsense mutations in a Becker
muscular dystrophy and an intermediate patient. _Hum Mutat_ 7: 72–75 Article CAS Google Scholar * Jarrett HW, Foster JL 1995 Alternative binding of actin and calmodulin to multiple sites
on dystrophin. _J Biol Chem_ 270: 5578–5586 Article CAS Google Scholar * Amann KJ, Renley BA, Ervasti JM 1998 A cluster of basic repeats in the dystrophin rod domain binds F-actin through
an electrostatic interaction. _J Biol Chem_ 273: 28419–28423 Article CAS Google Scholar * Pereboev AV, Ahmed N, thi Man N, Morris GE 2001 Epitopes in the interacting regions of
beta-dystroglycan (PPxY motif) and dystrophin (WW domain). _Biochem Biophys Acta_ 1527: 54–60 Article CAS Google Scholar * Suzuki A, Yoshida M, Hayashi K, Mizuno Y, Hagiwara Y, Ozawa E
1994 Molecular organization at the glycoprotein-complex-binding site of dystrophin. Three dystrophin-associated proteins bind directly to the carboxy-terminal portion of dystrophin. _Eur J
Biochem_ 220: 283–292 Article CAS Google Scholar * Yang B, Jung D, Rafael JA, Chamberlain JS, Campbell KP 1995 Identification of alpha-syntrophin binding to syntrophin triplet,
dystrophin, and utrophin. _J Biol Chem_ 270: 4975–4978 Article CAS Google Scholar * Blake DJ, Tinsley JM, Davies KE, Knight AE, Winder SJ, Kendrick-Jones J 1995 Coiled-coil regions in the
carboxy-terminal domains of dystrophin and related proteins: potentials for protein-protein interactions. _Trends Biochem Sci_ 20: 133–135 Article CAS Google Scholar * Sadoulet-Pucchio
HM, Rajala M, Kunkel LM 1997 Dystrobrevin and dystrophin: an interaction through coiled-coil motifs. _Proc Natl Acad Sci USA_ 94: 12413–12418 Article Google Scholar * Crawford GE, Faulkner
JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS 2000 Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. _J Cell Biol_ 150:
1399–1410 Article CAS Google Scholar * Kerst B, Mennerich D, Schuelke M, Stoltenburg-Didinger G, von Moers A, Gossrau R, van Landeghem FK, Speer A, Braun T, Hubner C 2000 Heterozygous
myogenic factor 6 mutation associated with myopathy and severe course of Becker muscular dystrophy. _Neuromuscul Disord_ 10: 572–577 Article CAS Google Scholar * Hattori N, Kaido M,
Nishigaki T, Inui K, Fujimura H, Nishimura T, Naka T, Hazama T 1999 Undetectable dystrophin can still result in a relatively benign phenotype of dystrophinopathy. _Neuromuscul Disord_ 9:
220–226 Article CAS Google Scholar * Davis DB, Delmonte AJ, Ly CT, McNally EM 2000 Myoferlin, a candidate gene and potential modifier of muscular dystrophy. _Hum Mol Genet_ 9: 217–226
Article CAS Google Scholar * Sicinski I, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ 1989 The molecular basis of muscular dystrophy in the mdx mouse: a point mutation.
_Science_ 244: 1578–1580 Article CAS Google Scholar Download references AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Pediatrics, Kobe University Graduate School of
Medicine, Kobe, 650-0017, Japan Ryo Suminaga, Yasuhiro Takeshima, Mariko Yagi & Masafumi Matsuo * Department of Pediatrics, Sakura Ryoikuen Hospital, Sanda, 669-1357, Japan Hiroko Wada
Authors * Ryo Suminaga View author publications You can also search for this author inPubMed Google Scholar * Yasuhiro Takeshima View author publications You can also search for this author
inPubMed Google Scholar * Hiroko Wada View author publications You can also search for this author inPubMed Google Scholar * Mariko Yagi View author publications You can also search for this
author inPubMed Google Scholar * Masafumi Matsuo View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Masafumi Matsuo.
ADDITIONAL INFORMATION This work was supported by grants from the Ministry of Education, Science and Culture of Japan and a Research Grant for Nervous and Mental Disorders from the Ministry
of Health and Welfare of Japan. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Suminaga, R., Takeshima, Y., Wada, H. _et al._ C-Terminal Truncated
Dystrophin Identified in Skeletal Muscle of an Asymptomatic Boy with a Novel Nonsense Mutation of the Dystrophin Gene. _Pediatr Res_ 56, 739–743 (2004).
https://doi.org/10.1203/01.PDR.0000142734.46609.43 Download citation * Received: 11 June 2003 * Accepted: 19 November 2003 * Issue Date: 01 November 2004 * DOI:
https://doi.org/10.1203/01.PDR.0000142734.46609.43 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link
is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative