Patient-derived ips cells for unveiling the molecular pathology of pelizaeus-merzbacher disease: a commentary on ‘reduced plp1 expression in induced pluripotent stem cells derived from a pelizaeus–merzbacher disease patient with a partial plp1 duplication’
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
In the studies of inherited disorders of the central nerve system (CNS), it is always challenging to determine how alterations in a disease-causing gene or a rearrangement of a genomic
segment that contains disease-causing genes can change gene expression and the resulting clinical phenotype. Conventional cell transfection assays employing expression plasmids cannot be
used when large genomic intervals that include multiple exons, introns, promoters, distant enhancer/suppressor elements or even multiple genes are involved. Lymphoblasts or fibroblasts
established from patients’ blood or skin can be examined, but only trace transcripts may be detectable for genes that are predominantly expressed in the CNS. In this issue of the _Journal of
Human Genetics_, Yamamoto and his colleagues have shown that induced pluripotent stem (iPS) cells derived from patients’ fibroblasts may serve as resources to directly determine gene
expression aberrations.1 They established iPS cells from three patients with Pelizaeus–Merzbacher disease (PMD), which is an X-linked disorder of the CNS. PMD is caused by mutations of the
_PLP1_ gene, which is predominantly expressed by oligodendrocytes, but is barely or faintly detectable in the lymphocytes or fibroblasts.2, 3 Of these three patients, one had a rare partial
duplication, encompassing a 16-Kb genomic interval that spans exons 1–3 of _PLP1_. In such a case, it is extremely difficult to determine the molecular outcome of the genomic rearrangement.
Yamamoto’s group resolved this problem by generating patient-derived iPS cells. They reported that _PLP1_ expression in iPS cells was 40 times higher than in fibroblasts. Therefore, it was
possible to directly visualize the _PLP1_ signals by northern blotting. Thus, undifferentiated iPS cells may serve as a better tool to examine _PLP1_ expression in patient-derived cells.
They showed that partial duplication of the gene completely diminished _PLP1_ expression, and this finding was compatible with the mild clinical presentation of this family. Meanwhile,
limitations in the use of undifferentiated iPS cells in the quantification of cryptic gene expression became apparent. It was expected that the iPS cells derived from a patient carrying a
>600-Kb duplication containing the entire _PLP1_ gene would show increased transcript levels. However, northern blot performed on three different cell lines showed a tendency only because
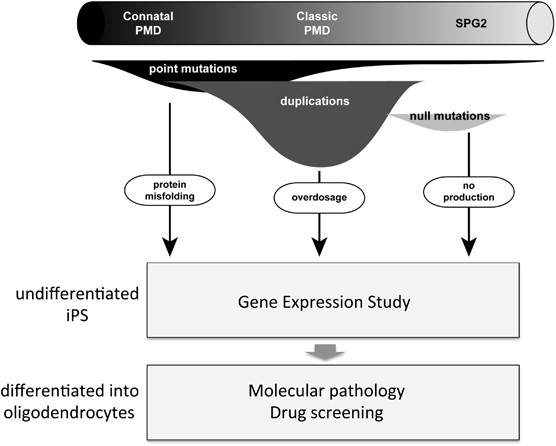
of the large experimental variation. iPS cells have greater potential than being a resource for gene expression studies. In PMD, various types of _PLP1_ mutations are associated with a
spectrum of clinical severities and the molecular mechanisms underlying each type of mutations are distinct (Figure 1).4 Differentiation of iPS cells into oligodendrocyte lineage cells will
enable modeling of the pathological process of different mutations in culture dishes. The potential treatment would differ according to the mutation type. For example, overexpression-induced
phenotypes could be mitigated by the downregulation of gene expression, whereas phenotypes arising from point mutations can be rescued via reduced endoplasmic reticulum stress.
Differentiation of iPS cells will also facilitate screening or monitoring of the effect of potential therapeutic reagents that could mitigate these cellular phenotypes. Meanwhile, terminal
differentiation of iPS cells into mature oligodendrocytes is both difficult and time consuming (requiring almost 3 months).5 In addition, these differentiated cells express mature myelin
proteins (for example, MBP and PLP1), but myelin-like membrane structures have not been reconstituted _in vitro_ thus far. Therefore, technical improvements in differentiation speed and
efficiency as well as _in vitro_ reconstitution of myelin is required to enable full use of these patient-derived iPS cells. REFERENCES * Shimojima, K., Inoue, T., Imai, Y., Arai, Y.,
Komoike, Y., Sugawara, M. _et al_. Reduced _PLP1_ expression in induced pluripotent stem cells derived from a Pelizaeus–Merzbacher disease patient with a partial PLP1 duplication. _J. Hum.
Genet._ 57, 580–586 (2012). Article CAS Google Scholar * Bonnet-Dupeyron, M. N., Combes, P., Santander, P., Cailloux, F., Boespflug-Tanguy, O. & Vaurs-Barrière, C. PLP1 splicing
abnormalities identified in Pelizaeus-Merzbacher disease and SPG2 fibroblasts are associated with different types of mutations. _Hum. Mutat_ 29, 1028–1036 (2008). Article CAS Google
Scholar * Regis, S., Grossi, S., Corsolini, F., Biancheri, R. & Filocamo, M. PLP1 gene duplication causes overexpression and alteration of the PLP/DM20 splicing balance in fibroblasts
from Pelizaeus-Merzbacher disease patients. _Biochim. Biophys. Acta_ 1792, 548–554 (2009). Article CAS Google Scholar * Inoue, K. PLP1-related inherited dysmyelinating disorders:
Pelizaeus-Merzbacher disease and spastic paraplegia type 2. _Neurogenetics_ 6, 1–16 (2005). Article CAS Google Scholar * Kang, S.-M., Cho, M. S., Seo, H., Yoon, C. J., Oh, S. K., Choi, Y.
M. _et al_. Efficient induction of oligodendrocytes from human embryonic stem cells. _Stem Cells_ 25, 419–424 (2007). Article CAS Google Scholar Download references AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * Department of Mental Retardation and Birth Defect Research, National Institute of Neuroscience, National Center of Neurology and Psychiatry, 4-1-1
Ogawahigashi-cho, Kodaira, 187-8502, Tokyo, Japan Ken Inoue Authors * Ken Inoue View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR
Correspondence to Ken Inoue. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Inoue, K. Patient-derived iPS cells for unveiling the molecular pathology of
Pelizaeus-Merzbacher disease: a commentary on ‘Reduced _PLP1_ expression in induced pluripotent stem cells derived from a Pelizaeus–Merzbacher disease patient with a partial _PLP1_
duplication’. _J Hum Genet_ 57, 553–554 (2012). https://doi.org/10.1038/jhg.2012.85 Download citation * Published: 12 July 2012 * Issue Date: September 2012 * DOI:
https://doi.org/10.1038/jhg.2012.85 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently
available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative