Congenital central hypoventilation syndrome with hyperinsulinism in a preterm infant
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Congenital central hypoventilation syndrome (CCHS), a rare disorder typically presenting in the newborn period, results in over 90% of cases from PHOX2B polyalanine repeat mutations. It is
characterized by alveolar hypoventilation, symptoms of autonomic nervous system dysregulation, and in a subset of cases Hirschsprung’s disease and, later, tumors of neural crest origin. We
describe a preterm infant with severe phenotype of CCHS and hyperinsulinism. A novel de novo heterozygote missence mutation (Gly68Cys) in the PHOX2B gene could be identified. Based on the
observation of three patients presenting with the combination of congenital hyperinsulinism and CCHS, hyperinsulinism might represent an additional clinical feature of CCHS.
Congenital central hypoventilation syndrome (CCHS) is a rare disorder with about 300 patients reported worldwide (Weese-Mayer and Berry-Kravis 2004). The syndrome comprises absence of
adequate control of respiration with decreased sensitivity to hypercapnia and hypoxia. Clinical onset occurs in the first few postnatal days in most patients, in the absence of neuromuscular
or lung disease or identifiable brain stem lesion. Infants with CCHS typically show adequate ventilation while awake, but hypoventilation with normal respiratory rates and diminished tidal
volume during sleep. More severely affected infants hypoventilate both when awake and asleep (Weese-Mayer et al. 1999). CCHS has been reported in association with aganglionic megacolon
(Hirschsprung’s disease) in ∼15–20% (Weese-Mayer and Berry-Kravis 2004; Croaker et al. 1998) and with tumors of neural crest origin: ganglioneuroma, neuroblastoma, and ganglioneuroblastoma,
in ∼5% of patients with CCHS (Rohrer et al. 2002).
CCHS also occurs with symptoms of diffuse autonomic nervous system dysregulation, including decreased heart rate variability, pupillary abnormality, esophageal dysmotility, decreased
perception of discomfort, sporadic profuse sweating episodes and decreased basal body temperature (Weese-Mayer and Berry-Kravis 2004). Mutations in the PHOX2B gene have been identified to be
the underlying genetic cause (Amiel et al. 2003; Matera et al. 2004). We report here the case of a preterm infant with CCHS and hyperinsulinism caused by a formerly unknown de novo mutation
in the PHOX2B gene.
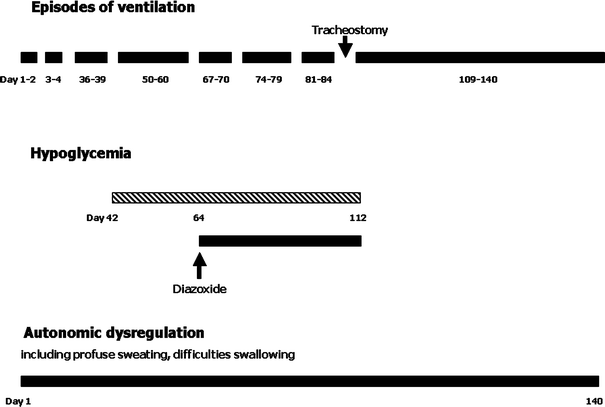
Graphic presentation of episodes of ventilation, hypoglycemia, and autonomic dysregulation including duration and the day of life of occurrence
Heterozygous transversion in exon 1 at position c.202G > T (the opposite strand is shown: C > A) results in a substitution from glycine to cysteine (Gly68Cys) in the PHOX2B protein
The combination of CCHS with Hirschsprung’s disease in an extremely preterm infant has been described previously (Bajaj et al. 2005). In our patient with profound and prolonged respiratory
insufficiency, no associated disease like Hirschsprung’s disease, that might give further clues towards CCHS, was existent. At a gestational age of 33 + 2 weeks, our patient did not
represent an extremely preterm infant, which made it difficult to assess whether intermittent respiratory insufficiency was still due to the condition of prematurity. The patient also showed
symptoms of autonomic nervous system dysregulation such as esophageal dysmotility. As premature infants often have problems like recurrent central apneas and feeding intolerance, diagnosing
CCHS in these patients is challenging. Furthermore, our patient presented with hyperinsulinism. Although congenital hyperinsulinism has been described in one patient with severe CCHS
(Meissner et al. 2001), hyperinsulinism is not considered a typical associated feature. In addition to the relative dysfunction in the enteric nervous system seen in CCHS, more complex
pathophysiological mechanisms may also result in persistent hyperinsulinism. As diffuse autonomic nervous system dysfunction is frequently seen in CCHS, pancreatic islets cells could also be
affected, which are richly innervated by parasympathetic, sympathetic and sensory nerves. Several different neurotransmitters are stored within the terminals of these nerves, both the
classical neurotransmitters, acetylcholine and noradrenaline, and several neuropeptides (Ahren 2000). We know from other investigations that PHOX2B delineates an uninterrupted chain of
sensors and neurons involved in the integration of central and peripheral chemoreception, including carotid bodies, chemoreceptor afferents, chemoresponsive projections to the ventrolateral
medulla, and central chemoreceptors (Stornetta et al. 2006). This is supported by both loss- and gain-of-function experiments which suggest that the transcription factor PHOX2B coordinates
quantitative and qualitative aspects of neurogenesis no matter if cholinergic, catecholaminergic, or serotonergic neurons are concerned (Dubreuil et al. 2000, 2002). Norepinephrine and
epinephrine-deficient mice have been reported to be hyperinsulinemic and to have lower blood glucose (Ste Marie and Palmiter 2003). These experiments have been performed in dopamine
beta-hydroxylase-null (Dbh−/−) mice. Knowing that dopamine beta-hydroxylase is co-expressed with PHOX2B would easily explain coexisting CCHS and hyperinsulinism with resulting hypoglycemia
(Trochet et al. 2005; Zhu et al. 2005). Numerous interactions of PHOX2B with other genes have been described: RET (Proto-oncogene tyrosine-protein kinase receptor ret precursor), GDNF (Glial
cell line-derived neurotrophic factor precursor), EDNRB (Endothelin B receptor precursor), ASCL1 (Achaete-scute homolog 1), DBH (Dopamine beta-hydroxylase precursor), TH (Tyrosine
3-monooxygena
se), NEUROG2 (Neurogenin 2), TLX3 (T-cell leukemia homeobox protein 3), SOX10 (Transcription factor SOX-10), and EDN3 (Endothelin-3 precursor). The closest interaction shown so far has been
described for dopamine beta-hydroxylase.
Furthermore, our patient is the third CCHS patient also presenting with hyperinsulinism. In both other patients (Meissner et al. 2001), a polyalanin repeat mutation was the underlying cause
(alanin expansion +6 and +7, respectively). Laboratory parameters on initial presentation were as follows: patient A: insulin 8.5 mU/l, glucose 1.8 mmol/l, ratio insulin/glucose: 4.7;
patient B: insulin 20.8 mU/l, glucose 2.0 mmol/l, ratio insulin/glucose: 10.4. Since different types of mutation are present in these three patients with CCHS and hyperinsulinism, this rare
clinical feature of CCHS is unlikely to be associated with either polyalanin or nonpolyalanin mutations. Regarding these three cases, it appears impossible to establish any kind of
correlation between extent and duration of hyperinsulinemia and the underlying mutation. However, hyperinsulinism may be linked to CCHS and could represent an additional clinical feature of
this complex disease.
CCHS without associated diseases like Hirschsprung’s disease is difficult to diagnose in preterm infants as a result of prematurity-associated apnea and bradycardia. Clinical suspicion,
additional symptoms like autonomic nervous system dysregulation, hyperinsulinism, and ultimately testing for PHOX2B mutation can confirm the diagnosis. Mutation testing may also help to
predict progression of the disease and allows genetic counselling of affected parents. Management of this rare disease presenting in a preterm infant is challenging due to the overlap with
specific problems of prematurity, the prognosis of CCHS presenting early in infancy is limited.
Anyone you share the following link with will be able to read this content: