Synthesis and structure–activity relationships of novel lincomycin derivatives. Part 4: synthesis of novel lincomycin analogs modified at the 6- and 7-positions and their potent antibacterial activities
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT To modify lincomycin (LCM) at the C-6 and the C-7 positions, we firstly prepared various substituted proline intermediates (7, 11–15 and 17). These proline intermediates were
coupled with methyl 1-thio-α-lincosamide and tetrakis-_O_-trimethylsilylation followed by selective deprotection of the TMS group at the 7-position gave a wide variety of key intermediates
(23–27, 47 and 50). Then, we synthesized a variety of novel LCM analogs modified at the 7-position in application of the Mitsunobu reaction, an SN2 reaction, and a Pd-catalyzed
cross-coupling reaction. Compounds 34 and 35 (1′-_N_H derivatives) exhibited enhanced antibacterial activities against resistant pathogens with _erm_ gene compared with the corresponding
1′-_N_-methyl derivatives (3 and 37). On the basis of reported SAR, we modified the 4′-position of LCM derivatives possessing a 5-(2-nitrophenyl)-1,3,4-thiadiazol-2-yl group at the C-7
position. Compound 56 showed significantly potent antibacterial activities against _S. pneumoniae_ and _S. pyogenes_ with _erm_ gene, and its activities against _S. pneumoniae_ with _erm_
gene were improved compared with those of 34 and 57. Although we synthesized novel analogs by transformation of a C-7 substituent focusing on the 1′-demethyl framework to prepare very potent
analogs 73 and 75, it was impossible to generate novel derivatives exhibiting stronger antibacterial activities against _S. pneumoniae_ with _erm_ gene compared with 56. SIMILAR CONTENT
BEING VIEWED BY OTHERS SYNTHETIC STUDIES ON THE TETRASUBSTITUTED D-RING OF CYSTOBACTAMIDS LEAD TO POTENT TEREPHTHALIC ACID ANTIBIOTICS Article Open access 05 November 2024 DESIGN, SYNTHESIS
AND ANTIBACTERIAL ACTIVITY OF NOVEL COLISTIN DERIVATIVES WITH THIOETHER BOND-MEDIATED CYCLIC SCAFFOLD Article 20 March 2023 DESIGN, SYNTHESIS AND OPTIMIZATION OF TARO INHIBITORS AS
MULTIFUNCTIONAL ANTIBIOTICS AGAINST METHICILLIN-RESISTANT _STAPHYLOCOCCUS AUREUS_ Article Open access 12 April 2025 INTRODUCTION Macrolide antibiotics have been used in the clinical site for
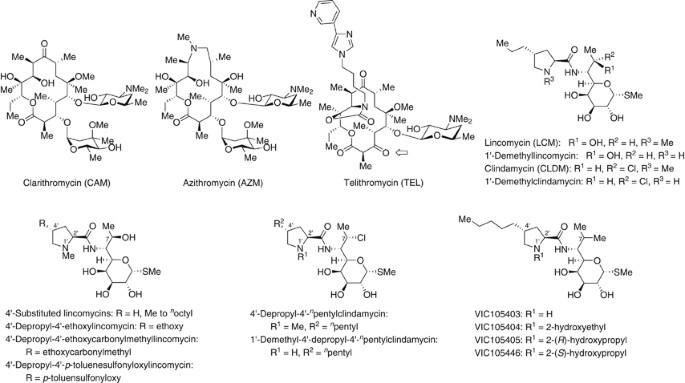
bacterial respiratory infections so far. Recently, macrolide-resistant bacteria with _erm_ gene have markedly increased.1, 2, 3 Clarithromycin (CAM)4 and azithromycin5, 6 are not effective
enough against resistant pathogens such as _Streptococcus pneumoniae_ and _Streptococcus pyogenes_ with _erm_ gene (Figure 1 and Table 1). Telithromycin (TEL)7 is effective enough against
_S. pneumoniae_ with _erm_ gene, but has the potential to cause a serious liver damage8, 9 and loss of consciousness.10, 11 Thus, TEL has scarcely been used in Japan. Novel azalides reported
by Miura _et al._12,13 are also effective against resistant pathogens, but these analogs are still under research process and have not been developed yet. Currently available oral
antibiotics are not effective enough against resistant bacteria with _erm_ and _mef_ genes causing respiratory infections and have some problems in safety or taste in clinical site.
Lincomycin (LCM)14, 15, 16, 17 and clindamycin (CLDM)18 are effective against pathogens with _mef_ gene in clinical isolates, but they are not effective against resistant pathogens with
_erm_ gene (Figure 1 and Table 1). As an overview, CLDM exhibits the following positive characters: (1) availability in _per os_ and intravenous administrations (switch therapy is possible),
(2) good distributions to tissue and cells, (3) suppression19 of toxin production by Streptococcal strains and (4) expected reasonable production cost of LCM/CLDM derivatives. Thus, LCM
derivatives might be more clinically valuable than macrolide antibiotics, if they are effective against pathogens with _erm_ gene. Chemical modifications at the C-7 position of LCM were
achieved by several research groups.17, 18, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32 None of them, however, showed effective antibacterial activities against resistant bacteria
with _erm_ gene. On the other hand, we reported 7(_S_)-thiolincomycin analogs exhibiting antibacterial activities against resistant pathogens with _erm_ gene as the first example33, 34, 35,
36 as far as we know. We recently reported 7(_S_)-thiolincomycin analogs, such as compounds 1–3 (Table 1), as the first-generation derivatives in our research.37 Those compounds possessed
weak antibacterial activities against resistant _S. pneumoniae_ with _erm_ and _mef_ genes, but compound 3 exhibited clearly improved activities compared with clarithromycin, azithromycin,
LCM and CLDM as shown in Table 1. Recently, we also reported 7-_S_-substituted novel LCM derivatives,38, 39, 40, 41 and some of them showed stronger activities than compound 3 did. Chemical
modification of a proline moiety at the C-6 position of LCM and CLDM was performed by several research groups.17, 24, 42, 43, 44, 45, 46, 47, 48 This portion has the following features: (1)
Regarding configuration between the 2′- and 4′-positions of a proline ring, an analog with _trans_ configuration exhibited more potent antibacterial activity compared with one with _cis_
configuration. (2) 4′-Depropyl-3′-propyllincomycin has not been synthesized, but 4′-depropyl-5′-propyllincomycin has already been synthesized. Its antibacterial activity was weaker than that
of LCM. (3) 1′-Demethylclindamycin (Figure 1) was two times as active _in vitro_ against _Sarcina lutea_ as CLDM, but 1′-demethyllincomycin was about one-twentieth as active as LCM
(relative potency against _S. lutea_: 1′-demethylclindamycin>CLDM>LCM>1′-demethyllincomycin=8>4>1>0.05).21, 42, 43 1′-Demethyl-1′-_N_-ethyllincomycin possessed the same
activity as LCM. (4) Regarding SAR of a chain length (H, Me to _n_-octyl) at the 4′-position of the proline moiety for LCM (Figure 1), the _in vitro_ antibacterial activities were enhanced
until reaching a maximum at the hexyl analog.21, 44 A similar _in vivo_ activity was indicated, but maximum effect was exhibited by an _n_-pentyl group. Furthermore, alternative _in vitro_
SAR were observed by changing a chain length (Figure 1) for a 4′-alkyl-substituent of CLDM and a 4′-alkyl-substituent of 1′-demethylclindamycin (relative potency:
4′-depropyl-4′-_n_-pentylclindamycin>CLDM, 1′-demethyl-4′-depropyl-4′-_n_-pentylclindamycin>1′-demethylclindamycin).21 However, antibacterial activities of those compounds against
resistant pathogens with _erm_ gene were not disclosed. (5) 4′-Depropyl-4′-ethoxylincomycin had only ~2% antibacterial activities of LCM, and 4′-depropyl-4′-ethoxycarbonylmethyllincomycin
and 4′-depropyl-4′-_p_-toluenesulfonyloxylincomycin were essentially inactive in antibacterial testing.17, 21, 45 On the other hand, Vicuron, which was consolidated by Pfizer, reported the
following compounds. VIC 105403 (Figure 1),24 possessing 1′-demethyl, 4′-_n_-pentyl and 7-methyl group, had potent activities compared with CLDM. VIC 105404,24 possessing
1′-_N_-(2-hydroxyethyl), 4′-_n_-pentyl and 7-methyl group, was already reported to have potent antibacterial activities as VIC 105403. However, those did not show antibacterial activities
against resistant pathogens with _erm_ gene. Furthermore, VIC 105405 and VIC 105446,24 having a branched side chain at the 1′-position, showed less antibacterial activities than CLDM. Other
VIC compounds possessing a branched side chain, a 3-(pyridin-4-yl)propyl or an _n_-butylthio group at the 4′-position, were already synthesized, but their antibacterial activities were not
disclosed in their patent.46 According to X-ray crystallographic analysis by Yonath _et al._,47 we hypothesized that CLDM had enough three-dimensional space around the 1′-position and it
might be able to enhance the antibacterial activities by filling the corresponding space with other functional groups except the Me group. On the basis of the above hypothesis, we designed
and synthesized novel 7(_S_)-substituted LCM derivatives modified at the 1′- and/or 4′-position(s) at the proline moiety. Anti-bacterial activities of these novel compounds are also
disclosed in this report. RESULTS AND DISCUSSION SYNTHESIS OF PROLINE DERIVATIVES Synthesis of proline derivatives is shown in Scheme 1. Synthetic route for modification of the proline
moiety at the 4′-position was already reported by Pedregal _et al._48, 49 We firstly prepared starting materials (4–6 and 16), and compound 8 was synthesized by hydroboration of 4 followed
by treatment with _tert_-butyldimethylsilyl chloride (TBSCl) or methyl iodide under the basic condition to give compounds 9 and 10. The compounds 4–6, 9, 10 and 16 were reduced with H2 in
the presence of Pd/C to give the corresponding carboxylic acids (7, 11–14 and 17), respectively. Compound 15 was prepared by hydrolysis of 6. 1H NMR spectra of compounds 7, 8, 10, 13–15 and
17 were observed as two sets of signals because of a rotamer by the _tert_-butoxycarbonyl (Boc) group. SYNTHESIS OF KEY INTERMEDIATES 23–27 AND TRANSFORMATION OF 23 AND 36 Synthesis of key
intermediates 23–27 and transformations of 23 and 36 are shown in Scheme 2. Compounds 18–22 were synthesized by condensation of compounds 7 and 12–15 with methyl 1-thio-α-lincosamide (MTL),
respectively. Tetra-_O_-trimethylsiliylation of all hydroxyl groups and successive regioselective deprotection of the TMS group at the 7-position gave key intermediates (23–27). Furthermore,
novel LCM derivatives 32–35 were synthesized via two or three steps from compound 23 by the Mitsunobu reaction at the 7-position and deprotection of the TMS groups and the Boc group at the
1′-position. On the other hand, 1′-_N_-methyl analog 37 was prepared from 36 following the similar procedure as the above. 1H NMR spectra of compounds 18–20 and 23 were observed as two sets
of signals because of a rotamer by the Boc group and compounds 25–27 showed broad peaks in 1H NMR spectra. Compounds 32–35 whose Boc groups were deprotected, however, showed a single set of
signals and sharp peaks in 1H NMR spectra. SYNTHESIS OF 1′-_N_-MODIFIED NOVEL LCM DERIVATIVES POSSESSING A SUBSTITUTED 1,3,4-THIADIAZOL-2-YLTHIO GROUP AT THE 7-POSITION Synthesis of
1′-_N_-modified novel LCM derivatives possessing the substituted 1,3,4-thiadiazol-2-ylthio group at the 7-position is shown in Scheme 3. Desired 1′-_N_-modified derivatives 38, 39, 41 and 45
were prepared from 34 or 35 by reductive aminoalkylation. Consequently, the TBS groups of 39 and 41 were removed by TBAF to give compounds 40 and 42, respectively. Compound 43 was
synthesized in application of (_R_)-2-methyloxirane under the basic condition from 35. Compound 44 was also prepared by acetic anhydride from 35. SYNTHESIS OF NOVEL LCM DERIVATIVES 48 AND 53
POSSESSING A GERMINAL BIS-PROPYL MOIETY OR A 3-(DIMETHYLAMINO)PROPYL GROUP AT THE 4′-POSITION Synthesis of novel LCM derivatives 48 and 53 possessing a germinal bis-propyl moiety and a
3-(dimethylamino)propyl group, respectively, at the 4′-position, and having the 5-(2-nitrophenyl)-1,3,4-thiadiazol-2-ylthio group at the 7-position, is shown in Scheme 4. Key intermediates
47 and 50 were, respectively, synthesized in the application of condensation, tetrakis-_O_-trimethylsilylation, and selective deprotection with the similar procedure to compound 23. Later, a
7-_S_-substituent was introduced to 47 by the Mitsunobu reaction, and deprotection gave a desired derivative 48. Compound 50 was transformed to 51 by the Mitsunobu reaction and
desilylation. Then, compound 51 was reacted with sodium azide, and an azide group was reduced to an amino group with triphenylphosphine-water. The afforded amino group in 52 was applied with
reductive aminoalkylation followed by deprotection of the Boc group under the acidic condition with trifluoroacetic acid (TFA) to give the desired novel derivative 53. 1H NMR spectra of
compounds 47 and 50 were also observed as broad peaks by the influence of a Boc group. Although the final product 53 could be purified as a single molecule, both intermediates 51 and 52
partially included impurity that was not removed by purification steps. SYNTHESIS OF NOVEL LCM DERIVATIVES WITH A VARIETY OF 4′-SUBSTITUENTS POSSESSING THE SUBSTITUTED
1,3,4-THIADIAZOL-2-YL-THIO GROUP AT THE 7-POSITION Synthesis of novel LCM derivatives having a 3-methoxypropyl, an _i_-butyl or an _n_-pentyl group at the 4′-position, whose 7-position was
the substituted 1,3,4-thiadiazol-2-yl-thio group, is shown in Scheme 5. Compounds 54–56 and 58 were prepared from key intermediates 24–26 by the similar procedure to compound 48. Reductive
aminoalkylation of 56 and 58 afforded the desired compounds 57 and 59, respectively. SYNTHESIS OF PROLINE-MODIFIED NOVEL LCM DERIVATIVES POSSESSING A (4-MORPHOLINOCARBONYL)PHENYLTHIO GROUP
AT THE 7-POSITION Synthesis of proline-modified novel LCM derivatives possessing a (4-morpholinocarbonyl)phenylthio group at the 7-position is shown in Scheme 6. Because LCM derivatives
possessing a (4-morpholinocarbonyl)phenylthio group at the 7-position exhibited improved antibacterial activities, we designed compounds 67–69. We already reported the methanesulfonyl (Ms)
route37, 38 to introduce a phenyl group via sulfur atom at the 7-position. The Ms route was applied to compounds 23 and 27 to give intermediates 60 and 61, respectively. 1H NMR spectra of
compounds 60–62 were observed as two sets of signals because of a rotamer by the Boc group. The desired analogs 67 and 68 were prepared from 60 and 62, respectively, by (i) hydrolysis under
the basic condition, (ii) condensation with morpholine and (iii) deprotection of the Boc group with TFA. Compound 69 was synthesized from 68 by reductive aminoalkylation with HCHO and
NaBH(OAc)3 in an acidic condition by AcOH. SYNTHESIS OF 73 AND 75 Synthesis of novel LCM derivatives 73 and 75 is shown in Scheme 7. We have already reported a Pd-catalyzed cross-coupling
route to introduce an aryl group at the 7-position of 7(_S_)-7-deoxy-7-mercaptlincomycin.40 A key intermediate 71 was prepared via four steps from 23, and precursors 72 and 74 were
synthesized by Pd-catalyzed cross-coupling reaction of 7140, 50 with the corresponding bromides. Deprotection of the Boc group afforded the desired analogs 73 and 75. 1H NMR spectra of
compounds 72 and 74 were observed as two sets of signals because of a rotamer by the Boc group, but both of the final compounds 73 and 75 showed a single set and sharp peaks in NMR spectra.
SAR ANALYSIS OF 7-_S_-SUBSTITUTED 1′-_N_H LCM DERIVATIVES (32–35) AND 1′-_N_-ME ANALOG 37 Antibacterial activity of LCM was reduced by 1′-_N_-demethylation, but that of CLDM was enhanced by
1′-_N_-demethylation. Thus, we were interested in the potency of 1′-_N_-demethyl products of 7(_S_)-substituted LCM derivatives 1–3 as shown in Table 1. Thus, we synthesized 1′-demethyl
analogs of 1–3 (32–34) and compounds 35 and 37 possessing an alternative 7-substituent. Antibacterial activities of those are shown in Table 2. Among them, 1′-_N_H derivatives 32, 34 and 35
exhibited improved antibacterial activities against _S. pneumoniae_ with _erm_ gene compared with the corresponding 1′-_N_-methyl analogs (1,3 and 37), respectively. Because 34 and 35
especially showed enhanced antibacterial activities against the target pathogens, we found that double modifications at the C-6 and C-7 positions were important to improve antibacterial
activities against _S. pneumoniae_ with _erm_ gene. SAR ANALYSIS OF ANTIBACTERIAL ACTIVITIES (MIC, ΜG ML−1) OF 7-_S_-SUBSTITUTED 1′-_N_-MODIFIED LCM DERIVATIVES (38, 40 AND 42–45) For the
purpose of accumulating detail information of SAR at the 1′-position, we synthesized novel LCM derivatives possessing various substituents at the 1′-position. At this point, the
5-(2-nitrophenyl)-1,3,4-thiadiazol-2-yl group and the 4-(5-methylamino-thiazole-4-yl)-1,3,4-thiadiazol-2-yl group were selected as a 7-substituent because of their SAR analysis (Tables 1 and
2). Consequently, compounds 40 and 42, possessing a 2-hydroxyethyl group at the 1′-position, showed anti-bacterial activities against target pathogens as shown in Table 3, but we concluded
that it was difficult to enhance antibacterial activities against _S. pneumoniae_ with _erm_ gene by introducing an alternative substituent at the 1′-position, except a hydrogen atom or a
methyl group. These results were closely related to SAR of 1′-_N_-alkyl-1′-demethyllincomycin.17, 21, 24 Then, we selected a hydrogen atom and a methyl group at the 1′-position for
modification of the proline moiety. SAR ANALYSIS OF 7-_S_-SUBSTITUTED 1′-_N_- AND 4′-MODIFIED LCM DERIVATIVES (48 AND 53–59) AND TEL To confirm whether 7-_S_-substituted LCM analogs show
similar anti-bacterial spectra to those reported previously, we synthesized 1′-demethyllincomycin derivatives possessing various substituents at the 4′-position. Structures of
7-_S_-substituents followed those in Table 3. As shown in Table 4, compounds 56 and 57, possessing an _n_-pentyl group instead of an _n_-propyl group at the 4′-position, exhibited strong
antibacterial activities against resistant bacteria with _erm_ gene. Although antibacterial activities of TEL against _S. pneumoniae_ with _erm_ gene were stronger than those of 56,
anti-bacterial activities of 56 against _S. pyogenes_ with _erm_ gene and resistant bacteria with _mef_ gene were remarkably stronger than those of TEL. SAR ANALYSIS OF 7-_S_-SUBSTITUTED
1′-_N_- AND 4′-MODIFIED LCM DERIVATIVES WITH AN ALTERNATIVE 7-_S_-SUBSTITUENT (67–69, 73 AND 75) We have reported significant potent antibacterial activities of LCM derivatives possessing a
substituted phenyl group at the C-7 position so far.40 This time, we transformed the proline moiety (the 1′-and 4′-position) of 7-_S_-substituted phenyl derivatives as shown in Table 5, such
as 7(_S_)-7-{4-(morphorinocarbonyl)phenylthio}lincomycin (76), 7(_S_)-7-{4-(pyridin-3-yl)phenylthio}lincomycin (77) and 7(_S_)-7-{4-(pyrimidin-5-yl)phenylthio}lincomycin (78). Their
antibacterial activities are shown in Table 5. The _n_-pentyl analogs 68 and 69 could not exhibit improved antibacterial activities against _S. pneumoniae_ and _S. pyogenes_ with _erm_ gene
compared with 56 and 57. On the other hand, 1′-_N_H LCM derivatives 73 and 75 with a 4-(pyridin-3-yl)phenyl group and a 4-(pyrimidin-5-yl)phenyl group, respectively, exhibited markedly
potent antibacterial activities against _S. pneumoniae_ with _erm_ gene. We confirmed that combination modification at the C-6 position (the proline moiety) and the C-7 position was
important to enhance antibacterial activities against _S. pneumoniae_ and _S. pyogenes_ with _erm_ gene. CONCLUSION To modify both the C-6 position (the proline moiety) and the C-7 position
of LCM, we firstly prepared various substituted proline intermediates. The intermediates were coupled with MTL to give a wide variety of 1′-_N_-Boc-1′-demethyllincomycin derivatives. The
7-_S_-substituents were introduced as follows. Key intermediates 23–26, 36, 47 and 50 were synthesized by the Mitsunobu reaction37, 38 with the corresponding thiol. Other key intermediates
23 and 27 were transformed to 7-_S_-benzoate by an SN2 reaction37, 38 via Ms derivatives, and the benzoate moiety was finally converted to a 4-(morpholinocarbonyl)phenyl moiety (Scheme 6).
7(_S_)-7-Deoxy-7-thiolincomycin (71) was coupled with biaryl bromide under the Pd-catalyzed cross-coupling reaction (Scheme 7).40, 50 Compounds 38, 40 and 42–45 were also prepared from 34 or
35. Those methodologies were found to be very practical to synthesize various LCM analogs modified at the C-6 and -7 positions. By SAR analysis of combination modification at the 1′- and
7-position, we concluded that it was difficult to enhance anti-bacterial activities against _S. pneumoniae_ with _erm_ gene by introducing an alternative substituent at the 1′-position,
except a hydrogen atom or a methyl group. Then, we selected a hydrogen atom and a methyl group at the 1′-position for further modification of the proline moiety. We next modified the
4′-position (in the proline moiety) of LCM derivatives possessing a 5-(2-nitrophenyl)-1,3,4-thiadiazol-2-yl group or a 4-(5-methylamino-thiazol-4-yl)-1,3,4-thiadiazol-2-yl group at the C-7
position (Table 4). Compounds 56 and 57, possessing an _n_-pentyl group instead of an _n_-propyl group at the 4′-position, exhibited strong antibacterial activities against resistant
bacteria with _erm_ gene. Although antibacterial activities of TEL against _S. pneumoniae_ with _erm_ gene were stronger than those of 56, antibacterial activities of 56 against _S.
pyogenes_ with _erm_ gene and resistant bacteria with _mef_ gene were remarkably stronger than those of TEL. We found that combination modification at the C-6 position (the proline moiety)
and the C-7 position was quite important to enhance antibacterial activities against _S. pneumoniae_ and _S. pyogenes_ with _erm_ gene. The above SAR might be partially related to the
polarity or water solubility of a molecule, but we only have limited SAR information. Further combination modification at the C-6 and C-7 positions of LCM analogs is in progress.
EXPERIMENTAL PROCEDURES GENERAL METHODS 1H NMR spectra were measured with a BRUKER Ascend 400 NMR spectrometer (Bruker Corporation, Coventry, UK) for 400 MHz, JEOL JNM-GSX 400 NMR
spectrometer (JEOL,Tokyo, Japan) for 400 MHz or a Varian Gemini 300 NMR spectrometer (Varian, Palo Alto, CA, USA) for 300 MHz in CDCl3 or CD3OD. TMS (0 p.p.m.) in CDCl3 or CD3OD was used as
an internal reference standard. Mass spectra (MS) were obtained on a JEOL JMS-700 mass spectrometer (JEOL) or Agilent Technologies 6530-Q-TOF LC/MS mass spectrometer (Agilent Technologies,
Santa Clara, CA, USA). The optical rotations were recorded with Jasco P-2300 digital polarimeter (Jasco Corporation, Tokyo, Japan). Column chromatography was performed with silica gel
(Wakogel C200; Wako Pure Chemical Industries, Osaka, Japan). Preparative thin layer chromatography was performed with silica gel (Merck, Darmstadt, Germany; TLC plates Silica gel 60 F254).
All organic extracts were dried over anhydrous MgSO4, and the solvent was removed with a rotary evaporator under reduced pressure.
(2_S_,4_R_)-1-_N_-(_TERT_-BUTOXYCARBONYL)-4-_N_-PROPYLPYRROLIDINE-2-CARBOXYLIC ACID (7) To a solution of compound 4 (1.0 g, 2.89 mmol) in MeOH (10 ml) was added Pd/C (100 mg), and then
vigorously stirred in hydrogen atmosphere at room temperature for 2 h. The mixture was filtrated with celite, and then the solution was concentrated under reduced pressure. The resulting
residue was filtrated with Chromatodisc (0.45 μm) (Kurabo Industries, Osaka, Japan). The filtrated solution was concentrated under reduced pressure to obtain the title compound (745 mg,
quant) as a colorless solid. FAB-MS _m/z_ 258 (M+H)+ as C13H23NO4; 1H NMR (400 MHz, CD3OD)) δ 0.70–0.93 (m, 3H), 1.10–1.41 (m, 13H), 1.65–1.87 (m, 1H), 1.93–2.06 (m, 1H), 2.10–2.27 (m, 1H),
2.73–2.89 (m, 1H), 3.43–3.63 (m, 1H) and 4.00–4.25 (m, 1H). (2_S_,4_R_)-2-BENZYL 1-_TERT_-BUTYL 4-(3-HYDROXYPROPYL)PYRROLIDINE-1,2-DICARBOXYLATE (8) To a solution of compound 4 (1.03 g, 2.98
mmol) in tetrahydrofuran (THF) (3 ml) was added 0.5 m 9-BBN in THF solution (8.95 ml, 4.47 mmol) and stirred at 50 °C for 1 h. Then, 1n NaOH (4 ml) and 35% H2O2 (4 ml) were added to the
mixture and stirred at 0 °C for 2 h. The solution was added to the saturated aqueous NaCl. The desired compound was extracted with ethyl acetate, and then the organic phase was dried over
Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=3/1 to 1/2) to obtain the title
compound (937 mg, 87%) as a colorless solid. EI-MS _m/z_ 363 (M)+ as C20H29NO5; 1H NMR (400 MHz, CDCl3) δ 1.31–1.48 (m, 11H), 1.49–1.60 (m, 2H), 1.75–1.92 (m, 1H), 2.05–2.14 (m, 1H),
2.19–2.39 (m, 1H), 2.88–3.05 (m, 1H), 3.57–3.83 (m, 3H), 4.25–4.50 (m, 1H), 4.96–5.32 (m, 2H) and 7.27–7.47 (m, 5H). (2_S_,4_R_)-2-BENZYL 1-_TERT_-BUTYL
4-(3-(_TERT_-BUTYLDIMETHYLSILYLOXY)PROPYL)PYRROLIDINE-1,2-DICARBOXYLATE (9) To a solution of compound 8 (2.80 g, 7.70 mmol) in dimethylformamide (DMF) (15 ml) were added imidazole (1.05 g,
15.4 mmol) and TBSCl (1.74 g, 11.56 mmol), and then stirred at room temperature for 30 min. The mixture was extracted with ethyl acetate and then the organic phase was dried over Na2SO4,
filtrated and concentrated under reduced pressure. The resulting residue was pumped up to obtain the title compound (3.50 g, crude). The total amount of this compound was used without
purification to synthesize 11. (2_S_,4_R_)-2-BENZYL 1-_TERT_-BUTYL 4-(3-METHOXYPROPYL)PYRROLIDINE-1,2-DICARBOXYLATE (10) To a solution of compound 8 (900 mg, 2.48 mmol) in DMF (9 ml) was
added 55% NaH in oil (99.2 mg, 3.72 mmol) and stirred at room temperature for 30 min. To the mixture was added methyl iodide (924 μl, 14.86 mmol) and then stirred at room temperature for 1
h. The solution was added to the saturated aqueous NaCl. The desired compound was extracted with ethyl acetate and then the organic phase was dried over Na2SO4, filtrated and concentrated
under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=20/1 to 4/1) to obtain the title compound (240 mg, 26%) as a colorless
oil. ESI-MS _m/z_ 378 (M+H)+ as C21H31NO5; 1H NMR (400 MHz, CDCl3) δ 1.33, 1.45 (s x 2, 9H), 1.35–1.50 (m, 2H), 1.50–1.57 (m, 2H), 1.75–1.91 (m, 1H), 2.03–2.13 (m, 1H), 2.18–2.33 (m, 1H),
2.90–3.04 (m, 1H), 3.31 (s, 3H), 3.25–3.40 (m, 2H), 3.59–3.80 (m, 1H), 4.25–4.47 (m, 1H), 5.03–5.29 (m, 2H) and 7.29–7.42 (m, 5H).
(2_S_,4_R_)-1-_N_-(_TERT_-BUTOXYCARBONYL)-4-(3-(_TERT_-BUTYLDIMETHYLSILYLOXY)PROPYL)PYRROLIDINE-2-CARBOXYLIC ACID (11) Compound 9 (3.50 g, crude) in MeOH (50 ml) were treated for 30 min
according to the similar procedure as described for the preparation of 7 to afford 11 (3.02 g, crude). The total amount of this compound was used to synthesize 49.
(2_S_,4_R_)-1-_N_-(_TERT_-BUTOXYCARBONYL)-4-(3-METHOXYPROPYL)PYRROLIDINE-2-CARBOXYLIC ACID (12) Compound 10 (200 mg, 0.530 mmol) in MeOH (2 ml) were treated for 1 h according to the similar
procedure as described for the preparation of 7 to afford 12 (152 mg, crude). The total amount of this compound was used without purification to synthesize 19.
(2_S_,4_R_)-1-_N_-(_TERT_-BUTOXYCARBONYL)-4-_I_-BUTYLPYRROLIDINE-2-CARBOXYLIC ACID (13) Compound 5 (195.3 mg, 0.54 mmol) in MeOH (2 ml) were treated for 30 min according to the similar
procedure as described for the preparation of 7 to afford 13 (141.1 mg, 96%) as an off-white solid. ESI-MS _m/z_ 272 (M+H)+ as C14H25NO4; 1H NMR (400 MHz, CD3OD) δ 0.90 (d, _J_=6.7 Hz, 3H),
0.92 (d, _J_=6.7 Hz, 3H), 1.23–1.35 (m, 2H), 1.41, 1.45 (s x 2, 9H), 1.51–1.64 (m, 1H), 1.76–1.95 (m, 1H), 2.07–2.15 (m, 1H), 2.28–2.44 (m, 1H), 2.85–3.00 (m, 1H), 3.57–3.74 (m, 1H) and
4.18–4.32 (m, 1H). (2_S_,4_R_)-1-_N_-(_TERT_-BUTOXYCARBONYL)-4-_N_-PENTYLPYRROLIDINE-2-CARBOXYLIC ACID (14) Compound 6 (1.69 g, 4.53 mmol) in MeOH (20 ml) were treated for 2 h according to
the similar procedure as described for the preparation of 7 to afford 14 (1.16 g, 89.5%) as a colorless solid. FAB-MS _m/z_ 286 (M+H)+ as C15H27NO4; 1H NMR (400 MHz, CD3OD) δ 0.80–0.98 (m,
3H), 1.20–1.51 (m, 8H), 1.42, 1.46 (s x 2, 9H), 1.78–1.98 (m, 1H), 2.10 (ddd, _J_=12.7, 6.2, 2.1 Hz, 1H), 2.19–2.36 (m, 1H), 2.85–3.01 (m, 1H), 3.57–3.75 (m, 1H), 4.17–4.33 (m, 1H).
(2_S_,4_R_)-1-_N_-(_TERT_-BUTOXYCARBONYL)-4-(_E_)-PENT-2-ENYL)PYRROLIDINE-2-CARBOXYLIC ACID (15) To a solution of compound 6 (2.0 g, 5.79 mmol) in MeOH (20 ml) was added 1 m aqueous NaOH (20
ml) and stirred at room temperature for 22 h. The mixture was diluted with H2O and Et2O and washed by Et2O. Aqueous layer was added to the saturated aqueous citric acid, extracted with
ethyl acetate and then the organic phase was washed with H2O, dried over Na2SO4, filtrated and concentrated under reduced pressure to obtain the title compound (1.55 g, 94.5%) as a colorless
solid. FAB-MS _m/z_ 284 (M+H)+ as C15H25NO4; 1H NMR (400 MHz, CD3OD) δ 0.89–1.05 (m, 3H), 1.41 (s, 6H), 1.45 (s, 3H), 1.85–2.15 (m, 6H), 2.23–2.39 (m, 1H), 2.92–3.09 (m, 1H), 3.52–3.68 (m,
1H), 4.18–4.31 (m, 1H), 5.30–5.44 (m, 1H) and 5.46–5.60 (m, 1H). 2(_S_)-1-_N_-(_TERT_-BUTOXYCARBONYL)-4,4-DI-_N_-PROPYLPYRROLIDINE-2-CARBOXYLIC ACID (17) Compound 16 (2.0 g, 5.19 mmol) in
MeOH (23 ml) were treated for 4.5 h according to the similar procedure as described for the preparation of 7 to afford 17 (1.55 g, quant) as an off-white oil. ESI-MS _m/z_ 300 (M+H)+ as
C16H29NO4; 1H NMR (400 MHz, CD3OD) δ 0.91 (t, _J_=6.9 Hz, 3H), 0.93 (t, _J_=6.9 Hz, 3H), 1.17–1.48 (m, 8H), 1.42, 1.45 (s x 2, 9H), 1.64–1.75 (m, 1H), 2.11–2.24 (m, 1H), 3.10 (d, _J_=10.6
Hz, 1H), 3.35–3.42 (m, 1H), 4.01–4.25 (m, 1H). 1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYLLINCOMYCIN (18) To a solution of compound 7 (745.0 mg, 2.90 mmol) in DMF (27 ml) were added
1-hydroxybenzotriazole (469.4 mg, 3.47 mmol), _N,N_′-dicyclohexylcarbodiimide (716.8 mg, 3.47 mmol) and MTL (1.10 g, 4.34 mmol) and stirred at room temperature for 15 h. The mixture was
added to H2O and ethyl acetate. The desired compound was extracted with ethyl acetate, and then the organic phase was washed with H2O, dried over Na2SO4, filtrated and concentrated under
reduced pressure to obtain the title compound. The total amount of this compound was used without purification to synthesize 23. For the qualified analytical purpose, the above crude
compound 18 was purified by column chromatography (ethyl acetate only) to obtain the title compound as a colorless solid. FAB-MS _m/z_ 493 (M+H)+ as C22H40N2O8S; 1H NMR (400 MHz, CD3OD) δ
0.78–0.93 (m, 3H), 1.05–1.45 (m, 7H), 1.35, 1.38 (s x 2, 9H), 1.59–1.81 (m, 1H), 1.88–2.09 (m, 1H), 1.97 (s, 3H), 2.23–2.43 (m, 1H), 2.83 (br t, _J_=10.1 Hz, 1H), 3.43–3.63 (m, 2H),
3.64–3.82 (m, 1H), 3.84–4.09 (m, 3H), 4.10–4.19 (m, 1H), 4.21–4.39 (m, 1H) and 5.14 (br d, _J_=5.4 Hz, 1H).
1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-DEPROPYL-4′-(3-METHOXYPROPYL)LINCOMYCIN (19) To a solution of compound 12 (152.3 mg, 0.53 mmol) in DMF (1.5 ml) were added
1-hydroxybenzotriazole (93.0 mg, 0.689 mmol), _N_,_N_′-dicyclohexylcarbodiimide (142.0 mg, 0.689 mmol) and MTL (174.0 mg, 0.689 mmol) and stirred at room temperature for 4.5 h. The mixture
was added to H2O and ethyl acetate. The desired compound was extracted with ethyl acetate, and then the organic phase was washed with H2O, dried over Na2SO4, filtrated and concentrated under
reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=1/1 to ethyl acetate only to ethyl acetate/MeOH=24/1) to obtain the title
compound (245.0 mg, 88.5% in two steps from 10) as a colorless solid. ESI-MS _m/z_ 523 (M+H)+ as C23H42N2O9S; 1H NMR (400 MHz, CD3OD) δ 1.15–1.29 (m, 3H), 1.44, 1.46 (s x 2, 9H), 1.36–1.66
(m, 4H), 1.73–1.88 (m, 1H), 2.06, 2.07 (s x 2, 3H), 2.09–2.17 (m, 1H), 2.33–2.50 (m, 1H), 2.93 (br t, _J_=9.9 Hz, 1H), 3.32 (s, 3H), 3.40 (t, _J_=6.3 Hz, 2H), 3.53–3.96 (m, 3H), 3.98–4.18
(m, 3H), 4.20–4.26 (m, 1H), 4.28–4.48 (m, 1H) and 5.24 (d, _J_=5.6 Hz, 1H). 1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-DEPROPYL-4′-_I_-BUTYLLINCOMYCIN (20) To a solution of compound 13
(57.9 mg, 0.21 mmol) in DMF (1.0 ml) were added 1-hydroxybenzotriazole (43.2 mg, 0.32 mmol), _N_,_N_′-dicyclohexylcarbodiimide (61.4 g, 0.32 mmol) and MTL (81.1 mg, 0.32 mmol) and stirred at
room temperature for 1 h. The mixture was added to the saturated aqueous NaHCO3 and ethyl acetate. The desired compound was extracted with ethyl acetate, and then the organic phase was
dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (chloroform/MeOH=10/1) to obtain the title
compound (100.2 mg, 92.7%) as a colorless solid. FAB-MS _m/z_ 507 (M+H)+ as C23H42N2O8S; 1H NMR (400 MHz, CD3OD) δ 0.87–0.98 (m, 6H), 1.16–1.32 (m, 6H), 1.44, 1.47 (s x 2, 9H), 1.51–1.66 (m,
1H), 1.70–1.86 (m, 1H), 2.05, 2.06 (s x 2, 3H), 2.07–2.15 (m, 1H), 2.39–2.57 (m, 1H), 2.90 (t, _J_=10.1 Hz, 1H), 3.55–3.71 (m, 1H), 3.72–3.88 (m, 1H), 3.98–4.19 (m, 2H), 4.23 (d, _J_=8.8
Hz, 1H), 4.32–4.51 (m, 1H) and 5.23 (d, _J_=5.4 Hz, 1H). 1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-DEPROPYL-4′-_N_-PENTYLLINCOMYCIN (21) Compound 14 (1.16 g, 4.05 mmol),
1-hydroxybenzotriazole (820 mg, 6.07 mmol), _N_,_N_′-dicyclohexylcarbodiimide (1.25 g, 6.07 mmol) and MTL (1.54 g, 6.07 mmol) in DMF (15.0 ml) were treated for 23 h according to the similar
procedure as described for the preparation of 18 to afford 21. The total amount of this compound was used without purification to synthesize 26.
1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-DEPROPYL-4′-{(_E_)-PENT-2-ENYL}LINCOMYCIN (22) Compound 15 (1.55 g, 5.47 mmol), 1-hydroxybenzotriazole (1.11 g, 8.21 mmol),
_N_,_N_′-dicyclohexylcarbodiimide (1.69 g, 8.21 mmol) and MTL (2.08 mg, 8.21 mmol) in DMF (23 ml) were treated for 3 h according to the similar procedure as described for the preparation of
18 to afford 22. The total amount of this compound was used without purification to synthesize 27. 1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-2,3,4-TRIS-_O_-(TRIMETHYLSILYL)LINCOMYCIN (23)
To a solution of compound 18 (crude) in pyridine (15 ml) were added trimethylchlorosilane (1.85 ml, 14.5 mmol) and hexamethyldisilazane (3.03 ml, 14.5 mmol) and stirred at room temperature
for 30 min, and then the solution was added to the saturated aqueous NaHCO3. The desired compound was extracted with ethyl acetate, washed with H2O and then the organic phase was dried over
Na2SO4, filtrated and concentrated under reduced pressure. To the resulting residue were added methanol (16.4 ml) and 6 n acetic acid (0.87 ml), and stirred at room temperature for 11 h. The
mixture was added to the saturated aqueous NaHCO3 and concentrated under reduced pressure to remove MeOH. The desired compound was extracted with ethyl acetate, and then the organic phase
was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1 to 1/2) to obtain
the title compound (1.45 g, 70.6% in three steps from 7) as a colorless solid. ESI-MS _m/z_ 709 (M+H)+ as C31H64N2O8SSi3; 1H NMR (400 MHz, CD3OD) δ 0.12–0.26 (m, 27H), 0.85–1.00 (m, 3H),
1.07–1.25 (m, 3H), 1.26–1.41 (m, 4H), 1.44, 1.46 (s x 2, 9H), 1.66–1.93 (m, 1H), 2.03, 2.05 (s x 2, 3H), 1.98–2.42 (m, 2H), 2.88–3.02 (m, 1H), 3.54–4.40 (m, 8H) and 5.18 (d, _J_=5.4 Hz, 1H).
1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-DEPROPYL-4′-(3-METHOXYPROPYL)-2,3,4-TRIS-_O_-(TRIMETHYLSILYL)LINCOMYCIN (24) Compound 19 (290 mg, 0.56 mmol), trimethylchlorosilane (355 μl,
2.77 mmol) and hexamethyldisilazane (581 μl, 2.77 mmol) in pyridine (1.0 ml) were treated for 1.0 h according to the similar procedure as described for the preparation of 23, and then the
crude compound and 6 n acetic acid (167 μl) in methanol (3.1 ml) were treated for 30 min according to the similar procedure as described for the preparation of 23 to afford 24 (282 mg, 68.7%
in two steps from 19) as a colorless solid. ESI-MS _m/z_ 739 (M+H)+ as C32H66N2O9SSi3.
1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-DEPROPYL-4′-_I_-BUTYL-2,3,4-TRIS-_O_-(TRIMETHYLSILYL)LINCOMYCIN (25) Compound 20 (652 mg, 1.63 mmol), trimethylchlorosilane (1.02 ml, 8.02 mmol)
and hexamethyldisilazane (1.68 ml, 8.02 mmol) in pyridine (3.5 ml) were treated for 1 h according to the similar procedure as described for the preparation of 23, and then the crude
compound and 6 n acetic acid (480 μl) in methanol (9 ml) were treated for 30 min according to the similar procedure as described for the preparation of 23 to afford 25 (946 mg, 79.8% in two
steps from 20) as a colorless solid. FAB-MS _m/z_ 723 (M+H)+ as C32H66N2O8SSi3; 1H NMR (400 MHz, CDCl3) δ 0.11–0.21 (m, 27H), 0.90 (d, _J_=6.6 Hz, 6H), 1.05–1.33 (m, 5H), 1.49 (s, 9H),
1.50–1.61 (m, 2H), 2.07 (s, 3H), 2.13–2.57 (m, 1H), 2.72–3.13 (m, 1H), 3.40–3.82 (m, 3H), 3.94–4.19 (m, 3H), 4.22–4.40 (m, 2H) and 5.19 (d, _J_=5.6 Hz, 1H).
1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-DEPROPYL-4′-_N_-PENTYL-2,3,4-TRIS-_O_-(TRIMETHYLSILYL)LINCOMYCIN (26) Compound 21 (crude), trimethylchlorosilane (2.6 ml, 20.3 mmol) and
hexamethyldisilazane (4.24 ml, 20.3 mmol) in pyridine (10.0 ml) were treated for 1 h according to the similar procedure as described for the preparation of 23, and then the crude compound
and 6 n acetic acid (1.21 ml) in methanol (23 ml) were treated for 2.0 h according to the similar procedure as described for the preparation of 23 to afford 26 (2.21 g, 74% in three steps
from 14) as a colorless solid. FAB-MS _m/z_ 737 (M+H)+ as C33H68N2O8SSi3; 1H NMR (400 MHz, CDCl3) δ 0.07–0.25 (m, 27H), 0.78–0.97 (m, 3H), 1.05–1.42 (m, 11H), 1.48 (s, 9H), 2.07 (s, 3H),
2.10–3.20 (m, 4H), 3.40–3.90 (m, 3H), 3.92–4.20 (m, 3H), 4.23–4.47 (m, 2H) and 5.19 (br d, _J_=5.4 Hz, 1H).
1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-DEPROPYL-4′-{(_E_)-PENT-2-ENYL}-2,3,4-TRIS-_O_-(TRIMETHYLSILYL)LINCOMYCIN (27) Compound 22 (crude), trimethylchlorosilane (3.50 ml, 27.4 mmol)
and hexamethyldisilazane (5.70 ml, 27.4 mmol) in pyridine (10 ml) were treated for 1 h according to the similar procedure as described for the preparation of 23 and then, the crude compound
and 6 n acetic acid (1.64 ml) in methanol (31 ml) were treated for 1.0 h according to the similar procedure as described for the preparation of 23 to afford 27 (3.29 g, 81.8% in three steps
from 15) as a colorless solid. 1H NMR (400 MHz, CDCl3) δ 0.02–0.28 (m, 27H), 0.96 (t, _J_=7.4 Hz, 3H), 1.08–1.25 (m, 3H), 1.48 (s, 9H), 1.89–2.51 (m, 6H), 2.02 (s, 3H), 2.66–3.26 (m, 2H),
3.40–3.64 (m, 2H), 3.68–4.19 (m, 4H), 4.22–4.50 (m, 2H), 5.19 (br d, _J_=5.1 Hz, 1H), 5.26–5.39 (m, 1H) and 5.43–5.59 (m, 1H).
7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-7-[5-{5-(METHYLAMINO)THIAZOL-4-YL}-1,3,4-THIADIAZOL-2-YLTHIO]-2,3,4-TRIS-_O_-(TRIMETHYLSILYL)LINCOMYCIN (31) To a solution of
compound 23 (200 mg, 0.28 mmol) in THF (2 ml) at 0 °C were added triphenylphosphine (110.9 mg, 0.42 mmol), diethylazodicarboxylate (77 μl, 0.42 mmol),
5-{5-(methylamino)thiazol-4-yl}-1,3,4-thiadiazole-2-thiol (100.7 mg, 0.44 mmol) and stirred at room temperature for 18 h. The solution was purified by preparative TLC (hexane/ethyl
acetate=2/1) to obtain the title compound as an off-white solid (88.8 mg, 34.2%). FAB-MS _m/z_ 921 (M+H)+ as C37H68N6O7S4Si3; 1H NMR (400 MHz, CDCl3) δ 0.00–0.25 (m, 27H), 0.80–1.00 (m, 3H),
1.08–1.67 (m, 16H), 1.74–2.99 (m, 7H), 3.02–3.22 (m, 3H), 3.43–3.90 (m, 3H), 3.98–4.50 (m, 4H), 4.60–4.94 (m, 1H), 5.20 (br d, _J_=5.4 Hz, 1H) and 7.85–8.00 (br s, 1H).
7(_S_)-7-(6-AMINOBENZOTHIAZOL-2-YLTHIO)-7-DEOXY-1′-DEMETHYLLINCOMYCIN (32) To a solution of compound 23 (200 mg, 0.28 mmol) in THF (2 ml) at 0 °C were added triphenylphosphine (110.9 mg,
0.42 mmol), diethylazodicarboxylate (77 μl, 0.42 mmol), 6-aminobenzothiazole-2-thiol (79.7 mg, 0.44 mmol) and stirred at room temperature for 4 h. To the solution was added 1 n HCl (1
ml)–MeOH (1 ml) and stirred at room temperature for 30 min. The solution was added to the saturated aqueous NaHCO3. The desired compound was extracted with ethyl acetate, and then the
organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain
7(_S_)-7-(6-aminobenzothiazol-2-yl)thio-1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-7-deoxy-2,3,4-tris-_O_-(trimethylsilyl)lincomycin (197.2 mg, crude). To the solution of this intermediate
in MeOH (2 ml) was added 4 n HCl-ethyl acetate (2.5 ml) and stirred at room temperature for 2 h. The solution was concentrated under reduced pressure. The resulting residue was purified by
silica gel column chromatography (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound (96.4 mg, 61.4% in three steps from 23) as an off-white solid. [α]D26+92.1° (_c_ 2.49, MeOH);
ESI-MS _m/z_ 557 (M+H)+ as C24H36N4O5S3; TOF-ESI-HRMS (M+H)+ calcd. for C24H36N4O5S3: 557.1926, found: 557.1920; 1H NMR (400 MHz, CD3OD) δ 0.86–0.96 (m, 3H), 1.25–1.40 (m, 4H), 1.49 (d,
_J_=6.9 Hz, 3H), 1.69–1.82 (m, 1H), 1.93 (s, 3H), 1.96–2.13 (m, 2H), 2.52 (dd, _J_=10.4, 8.1 Hz, 1H), 3.20 (dd, _J_=10.4, 6.9 Hz, 1H), 3.58 (dd, _J_=10.2, 3.2 Hz, 1H), 3.80–3.87 (m, 2H),
4.11 (dd, _J_=10.2, 5.6 Hz, 1H), 4.27 (dq, _J_=6.9, 2.7 Hz, 1H), 4.39 (br dd, _J_=10.0, 0.9 Hz, 1H), 4.57 (dd, _J_=10.0, 2.7 Hz, 1H), 5.26 (d, _J_=5.6 Hz, 1H), 6.85 (dd, _J_=8.7, 2.1 Hz,
1H), 7.08 (d, _J_=2.1 Hz, 1H) and 7.59 (d, _J_=8.7 Hz, 1H). 7(_S_)-7-(5-AMINO-1,3,4-THIADIAZOL-2-YLTHIO)-7-DEOXY-1′-DEMETHYLLINCOMYCIN (33) To a solution of compound 23 (200 mg, 0.28 mmol)
in THF (2 ml) at 0 °C were added triphenylphosphine (138.0 mg, 0.53 mmol), diethylazodicarboxylate (96 μl, 0.53 mmol), 5-(_tert_-butoxycarbonylamino)-1,3,4-thiadiazole-2-thiol (126.9 mg,
0.54 mmol) and stirred at room temperature for 3 h. To the solution was added 1 n HCl (1 ml)–MeOH (1 ml), and stirred at room temperature for 50 min. The solution was added to the saturated
aqueous NaHCO3. The desired compound was extracted with ethyl acetate, and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting
residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain
7(_S_)-7-[5-{(_tert_-butoxycarbonyl)amino}-1,3,4-thiadiazol-2-ylthio]-1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-7-deoxylincomycin as a colorless solid (146.6 mg, 73.4%). 1H NMR (400 MHz,
CD3OD) δ 0.85–0.98 (m, 3H), 1.25–1.60 (m, 25 H), 1.80–1.97 (m, 1H), 1.98–2.17 (m, 1H), 2.06, 2.10 (s x 2, 3H), 2.22–2.40 (m, 1H), 2.89–3.05 (m, 1H), 3.50–3.60 (m, 1H), 3.69–3.82 (m, 1H),
3.84–4.00 (m, 1H), 4.02–4.20 (m, 2H), 4.26–4.42 (m, 2H), 4.45–4.60 (m, 1H) and 5.27 (br d, _J_=5.4 Hz, 1H). To the solution of this intermediate (146.6 mg, 0.21 mmol) in MeOH (1.4 ml) was
added 4 n HCl-ethyl acetate (1.7 ml), and stirred at room temperature for 2 h. The solution was concentrated under reduced pressure. The resulting residue was purified by preparative TLC
(CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound (25.4 mg, 24.2%) as a colorless solid. [α]D25 +101° (_c_ 0.33, MeOH); ESI-MS _m/z_ 508 (M+H)+ as C19H33N5O5S3; TOF-ESI-HRMS
(M+H)+ calcd. for C19H33N5O5S3: 508.1722, found: 508.1719; 1H NMR (400 MHz, CD3OD) δ 0.89–0.98 (m, 3H), 1.33–1.45 (m, 4H), 1.37 (d, _J_=7.0 Hz, 3H), 1.84–1.95 (m, 1H), 2.03–2.18 (m, 2H),
2.11 (s, 3H), 2.66 (dd, _J_=10.6, 8.3 Hz, 1H), 3.30–3.36 (m, 1H), 3.56 (dd, _J_=10.2, 3.2 Hz, 1H), 3.81 (br dd, _J_=3.2, 0.7 Hz, 1H), 3.96 (dq, _J_=7.0, 2.6 Hz, 1H), 4.00 (dd, _J_=9.3, 4.0
Hz, 1H), 4.09 (dd, _J_=10.2, 5.6 Hz, 1H), 4.37 (br dd, _J_=10.0, 0.7 Hz, 1H), 4.52 (dd, _J_=10.0, 2.6 Hz, 1H) and 5.27 (d, _J_=5.6 Hz, 1H).
7(_S_)-1′-DEMETHYL-7-DEOXY-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YLTHIO}LINCOMYCIN (34) To a solution of compound 23 (200 mg, 0.28 mmol) in THF (2 ml) at 0 °C were added triphenylphosphine
(110.9 mg, 0.42 mmol), diethylazodicarboxylate (77 μl, 0.42 mmol), 5-(2-nitrophenyl)-1,3,4-thiadiazole-2-thiol (104.6 mg, 0.44 mmol), and stirred at room temperature for 7 h. The solution
was added to the saturated aqueous NaHCO3. The desired compound was extracted with ethyl acetate, and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced
pressure. To the resulting residue was added MeOH (4 ml)–1n HCl (1 ml) and stirred at room temperature for 2.5 h. The solution was added to the saturated aqueous NaHCO3. The desired compound
was extracted with ethyl acetate, and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by preparative TLC
(CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain 7(_S_)-1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-7-deoxy-7-{5-(2-nitrophenyl)-1,3,4-thiadiazol-2-ylthio}lincomycin (164.9 mg as crude). To the
solution of this crude compound (63.9 mg) in MeOH (0.6 ml) was added 4 n HCl-ethyl acetate (0.75 ml), and stirred at room temperature for 2.5 h. The solution was concentrated under reduced
pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound (13.0 mg, 19.4% in three steps from 23) as a colorless solid.
[α]D26 +37.1° (_c_ 0.21, MeOH); ESI-MS _m/z_ 614 (M+H)+ as C25H35N5O7S3; TOF-ESI-HRMS (M+H)+ calcd. for C25H35N5O7S3: 614.1777, found: 614.1778; 1H NMR (400 MHz, CD3OD) δ 0.83–0.99 (m, 3H),
1.30–1.45 (m, 4H), 1.56 (d, _J_=7.0 Hz, 3H), 1.88–1.98 (m, 1H), 1.99 (s, 3H), 2.07–2.25 (m, 2H), 2.69 (br dd, _J_=10.6, 8.4 Hz, 1H), 3.32–3.42 (m, 1H), 3.56 (dd, _J_=10.2, 3.2 Hz, 1H),
3.82–3.88 (m, 1H), 4.00–4.10 (m, 1H), 4.10 (dd, _J_=10.2, 5.6 Hz, 1H), 4.36–4.46 (m, 1H), 4.47 (dq, _J_=7.0, 2.5 Hz, 1H), 4.66 (dd, _J_=10.0, 2.5 Hz, 1H), 5.28 (d, _J_=5.6 Hz, 1H), 7.74–7.87
(m, 3H) and 8.05–8.15 (m, 1H). 7(_S_)-1′-DEMETHYL-7-DEOXY-7-[5-{5-(METHYLAMINO)THIAZOL-4-YL}-1,3,4-THIADIAZOL-2-YLTHIO]LINCOMYCIN (35) To the solution of compound 31 (88.8 mg 0.096 mmol) in
MeOH (0.5 ml) was added 4 n HCl-ethyl acetate (0.79 ml), stirred at 0 °C for 1 h and then stirred at room temperature for 3.5 h. The solution was concentrated under reduced pressure. The
resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound (39.8 mg, 68.3%) as an off-white solid. [α]D26 +69.5° (_c_ 0.60, MeOH);
ESI-MS _m/z_ 605 (M+H)+ as C23H36N6O5S4; TOF-ESI-HRMS (M+H)+ calcd. for C23H36N6O5S4: 605.1708, found: 605.1706; 1H NMR (400 MHz, CD3OD) δ 0.86–0.95 (m, 3H), 1.29–1.41 (m, 4H), 1.49 (d,
_J_=6.9 Hz, 3H), 1.78–1.87 (m, 1H), 1.98–2.14 (m, 2H), 2.01 (s, 3H), 2.56 (dd, _J_=10.5, 8.1 Hz, 1H), 3.11 (s, 3H), 3.25 (dd, _J_=10.5, 7.0 Hz, 1H), 3.56 (dd, _J_=10.3, 3.2 Hz, 1H), 3.82 (br
dd, _J_=3.2, 0.9 Hz, 1H), 3.86 (dd, _J_=9.2, 3.9 Hz, 1H), 4.09 (dd, _J_=10.3, 5.6 Hz, 1H), 4.27 (dq, _J_=6.9, 2.7 Hz, 1H), 4.39 (br dd, _J_=10.0, 0.9 Hz, 1H), 4.59 (dd, _J_=10.0, 2.7 Hz,
1H), 5.26 (d, _J_=5.6 Hz, 1H) and 8.12 (s, 1H). 7(_S_)-7-DEOXY-7-[5-{5-(METHYLAMINO)THIAZOL-4-YL}-1,3,4-THIADIAZOL-2-YLTHIO]LINCOMYCIN (37) Compound 36 (240 mg, 0.39 mmol),
triphenylphosphine (150.0 mg, 0.57 mmol), diethylazodicarboxylate (100 μl, 0.64 mmol) and 5-{5-(methylamino)thiazol-4-yl}-1,3,4-thiadiazole-2-thiol (150.0 mg, 0.65 mmol) in THF (5 ml) were
treated for 2 h, and then to the solution was added 1n HCl (0.5 ml)–MeOH (5 ml) and stirred at room temperature for 1 h. The solution was then added to the saturated aqueous NaHCO3. The
desired compound was extracted with ethyl acetate, and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by
preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound 37 (88.2 mg, 37% in two steps) as an off-white solid. [α]D26 +125° (_c_ 0.89, MeOH); ESI-MS _m/z_ 619 (M+H)+
as C24H38N6O5S4; TOF-ESI-HRMS (M+H)+ calcd. for C24H38N6O5S4: 619.1865, found: 619.1860; 1H NMR (400 MHz, CD3OD) δ 0.82–0.98 (m, 3H), 1.21–1.39 (m, 4H), 1.53 (d, _J_=6.9 Hz, 3H), 1.78–1.89
(m, 1H), 1.92–2.08 (m, 2H), 2.02 (s, 3H), 2.09–2.25 (m, 1H), 2.35 (s, 3H), 3.00 (dd, _J_=10.5, 5.0 Hz, 1H), 3.13 (s, 3H), 3.19 (dd, _J_=8.5, 6.2 Hz, 1H), 3.59 (dd, _J_=10.2, 3.2 Hz, 1H),
3.77–3.85 (m, 1H), 4.11 (dd, _J_=10.2, 5.6 Hz, 1H), 4.27 (dq, _J_=6.9, 3.0 Hz, 1H), 4.44 (br dd, _J_=9.8, 0.5 Hz, 1H), 4.57 (dd, _J_=9.8, 3.0 Hz, 1H), 5.27 (d, _J_=5.6 Hz, 1H) and 8.13 (s,
1H). 7(_S_)-1′-DEMETHYL-7-DEOXY-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YL}THIO-1′-_N_-_I_-PROPYLLINCOMYCIN (38) To a solution of compound 34 (30.5 mg, 0.05 mmol) in 1,2-dichloroethane (1
ml) at 0 °C were added acetone (40 μl, 0.054 mmol), AcOH (one drop) and NaBH(OAc)3 (21.7 mg, 0.10 mmol) and stirred at room temperature for 15 h. The mixture was concentrated under reduced
pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq NH4OH=10/1/0.1) and then by LH-20 (CHCl3/MeOH=1/1) to obtain the title compound (20.1 mg, 61%) as a
colorless solid. [α]D26 +76.8° (_c_ 0.59, MeOH); ESI-MS _m/z_ 656 (M+H)+ as C28H41N5O7S3; TOF-ESI-HRMS (M+H)+ calcd. for C28H41N5O7S3: 656.2246, found: 656.2243; 1H NMR (400 MHz, CD3OD) δ
0.86–0.98 (m, 3H), 1.06–1.16 (m, 6H), 1.28–1.42 (m, 4H), 1.58 (d, _J_=6.9 Hz, 3H), 1.69–1.81 (m, 1H), 1.99 (s, 3H), 1.97–2.07 (m, 1H), 2.07–2.20 (m, 1H), 2.20–2.31 (m, 1H), 2.76–2.90 (m,
1H), 3.26–3.34 (m, 1H), 3.39–3.52 (m, 1H), 3.57 (dd, _J_=10.2, 3.2 Hz, 1H), 3.82 (br dd, _J_=3.2, 0.8 Hz, 1H), 4.10 (dd, _J_=10.2, 5.6 Hz, 1H), 4.40 (br dd, _J_=9.3, 0.8 Hz, 1H), 4.51 (dq,
_J_=6.9, 3.4 Hz, 1H), 4.59 (dd, _J_=9.3, 3.4 Hz, 1H), 5.26 (d, _J_=5.6 Hz, 1H), 7.74–7.86 (m, 3H) and 8.06–8.12 (m, 1H).
7(_S_)-1′-_N_-{2-(_TERT_-BUTYLDIMETHYLSILYLOXY)ETHYL}-1′-DEMETHYL-7-DEOXY-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YLTHIO}LINCOMYCIN (39) Compound 34 (49.2 mg, 0.08 mmol),
2-(_tert_-butyldimethylsilyloxy)acetaldehyde (23 μl, 0.12 mmol), AcOH (one drop) and NaBH(OAc)3 (34.2 mg, 0.16 mmol) in 1,2-dichloroethane (1 ml) were treated at 0 °C for 15 h according to
the similar procedure as described for the preparation of 38 to afford 39 (26.9 mg, 44.0%) as a colorless solid. FAB-MS _m/z_ 772 (M+H)+ as C33H53N5O8S3Si; 1H NMR (400 MHz, CD3OD) δ 0.07 (s,
3H), 0.08 (s, 3H), 0.82–0.98 (m, 12H), 1.25–1.42 (m, 4H), 1.59 (d, _J_=6.8 Hz, 1H), 1.68–1.87 (m, 1H), 1.94–2.05 (m, 1H), 1.97 (s, 3H), 2.07–2.21 (m, 2H), 2.54–2.92 (m, 2H), 3.37–3.44 (m,
1H), 3.56 (dd, _J_=10.2, 3.2 Hz, 1H), 3.71–3.92 (m, 4H), 4.10 (dd, _J_=10.2, 5.6 Hz, 1H), 4.35–4.44 (m, 1H), 4.47–4.56 (m, 1H), 4.58–4.65 (m, 1H), 5.26 (d, _J_=5.6 Hz, 1H), 7.74–7.86 (m, 3H)
and 8.06–8.13 (m, 1H). 7(_S_)-1′-DEMETHYL-7-DEOXY-1′-_N_-(2-HYDROXYETHYL)-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YLTHIO}LINCOMYCIN (40) To a solution of compound 39 (26.9 mg, 0.035 mmol)
in THF (0.5 ml) at 0 °C were added 1m THF solution of _tetra_-_n_-butyl ammonium fluoride (100 μl, 0.10 mmol) and stirred at room temperature for 15 h. The mixture was concentrated under
reduced pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq NH4OH=10/1/0.1) to obtain the title compound (16.5 mg, 72%) as a colorless solid. [α]D25 +70.5°
(_c_ 0.22, MeOH); ESI-MS _m/z_ 658 (M+H)+ as C27H39N5O8S3; TOF-ESI-HRMS (M+H)+ calcd. for C27H39N5O8S3: 658.2039, found: 658.2044; 1H NMR (400 MHz, CD3OD) δ 0.87–0.96 (m, 3H), 1.29–1.43 (m,
4H), 1.60 (d, _J_=6.9 Hz, 3H), 1.79–1.93 (m, 1H), 1.99 (s, 3H), 2.02–2.10 (m, 1H), 2.10–2.23 (m, 2H), 2.64–2.75 (m, 1H), 2.83–2.96 (m, 1H), 3.31–3.40 (m, 1H), 3.40–3.50 (m, 1H), 3.56 (dd,
_J_=10.3, 3.2 Hz, 1H), 3.63–3.77 (m, 2H), 3.83 (dd, _J_=3.2, 0.8 Hz, 1H), 4.10 (dd, _J_=10.3, 5.6 Hz, 1H), 4.45 (br dd, _J_=9.9, 0.8 Hz, 1H), 4.51 (dq, _J_=6.9, 2.9 Hz, 1H), 4.63 (dd,
_J_=9.9, 2.9 Hz, 1H), 5.26 (d, _J_=5.6 Hz, 1H), 7.74–7.86 (m, 3H) and 8.06–8.12 (m, 1H).
7(_S_)-1′-_N_-{2-(_TERT_-BUTYLDIMETHYLSILYLOXY)ETHYL}-1′-DEMETHYL-7-DEOXY-7-[5-{5-(METHYLAMINO)THIAZOL-4-YL}-1,3,4-THIADIAZOL-2-YLTHIO]LINCOMYCIN (41) Compound 35 (74.3 mg, 0.12 mmol),
2-(_tert_-butyldimethylsilyloxy)acetaldehyde (34 μl, 0.18 mmol), AcOH (one drop) and NaBH(OAc)3 (51.0 mg, 0.24 mmol) in 1,2-dichloroethane (1 ml) were treated at room temperature according
to the similar procedure as described for the preparation of 38 to afford 41 (54.8 mg, 60%) as a colorless solid. FAB-MS _m/z_ 763 (M+H)+ as C31H54N6O6S4Si; 1H NMR (400 MHz, CD3OD) δ 0.05
(s, 3H), 0.06 (s, 3H), 0.78–1.00 (m, 12H), 1.23–1.42 (m, 4H), 1.54 (d, _J_=7.1 Hz, 3H), 1.72–1.86 (m, 1H), 1.92–2.05 (m, 1H), 1.99 (s, 3H), 2.08–2.22 (m, 2H), 2.50–2.87 (m, 2H), 3.12 (s,
3H), 3.25–3.30 (m, 1H), 3.36–3.44 (m, 1H), 3.52–3.60 (m, 1H), 3.68–3.88 (m, 3H), 4.10 (dd, _J_=10.2, 5.6 Hz, 1H), 4.30–4.44 (m, 2H), 4.54–4.62 (m, 1H), 5.26 (d, _J_=5.6 Hz, 1H) and 8.13 (s,
1H). 7(_S_)-1′-DEMETHYL-7-DEOXY-1′-_N_-(2-HYDROXYLETHYL)-7-[5-{5-(METHYLAMINO)THIAZOL-4-YL}-1,3,4-THIADIAZOL-2-YLTHIO]LINCOMYCIN (42) Compound 41 (54.8 mg, 0.072 mmol) and 1 m THF solution
of _tetra_-_n_-butyl ammonium fluoride (200 μl, 0.20 mmol) in THF (1.0 ml) were treated at 0 °C for 1 h and then treated at room temperature for 5 h according to the similar procedure as
described for the preparation of 40 to afford 42 (31.3 mg, 67.0%) as a colorless solid. [α]D25 +51.1° (_c_ 0.27, MeOH); ESI-MS _m/z_ 649 (M+H)+ as C25H40N6O6S4; TOF-ESI-HRMS (M+H)+ calcd.
for C25H40N6O6S4: 649.1970, found: 649.1973; 1H NMR (400 MHz, CD3OD) δ 0.88–0.96 (m, 3H), 1.30–1.40 (m, 4H), 1.54 (d, _J_=6.9 Hz, 3H), 1.79–1.90 (m, 1H), 1.97–2.08 (m, 1H), 2.00 (s, 3H),
2.09–2.21 (m, 2H), 2.62–2.72 (m, 1H), 2.83–2.93 (m, 1H), 3.11 (s, 3H), 3.30–3.37 (m, 1H), 3.39–3.45 (m, 1H), 3.56 (dd, _J_=10.2, 3.2 Hz, 1H), 3.65–3.74 (m, 2H), 3.81 (br dd, _J_=3.2, 0.8 Hz,
1H), 4.09 (dd, _J_=10.2, 5.6 Hz, 1H), 4.33 (dq, _J_=6.9, 2.8 Hz, 1H), 4.43 (br dd, _J_=9.8, 0.8 Hz, 1H), 4.59 (dd, _J_=9.8, 2.8 Hz, 1H), 5.25 (d, _J_=5.6 Hz, 1H) and 8.11 (s, 1H).
7(_S_)-1′-DEMETHYL-7-DEOXY-1′-_N_-{2(_R_)-HYDROXYPROPYL}-7-[5-{5-(METHYLAMINO)THIAZOL-4-YL}-1,3,4-THIADIAZOL-2-YLTHIO]LINCOMYCIN (43) To a solution of compound 35 (28.7 mg, 0.048 mmol) and
_N,N_-diisopropylethylamine (10.0 μl, 0.057 mmol) in MeOH (1 ml) at 0 °C was added (_R_)-2-methyloxirane (0.30 ml, 4.3 mmol) and stirred at 0 °C for 96 h. The mixture was concentrated under
reduced pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=10/1/0.1) to obtain the title compound (10.6 mg, 33%) as a colorless solid. [α]D26 +29.2°
(_c_ 0.15, MeOH); ESI-MS _m/z_ 663 (M+H)+ as C26H42N6O6S4; TOF-ESI-HRMS (M+H)+ calcd. for C26H42N6O6S4: 663.2127, found: 663.2127; 1H NMR (400 MHz, CD3OD) δ 0.87–0.96 (m, 3H), 1.16 (d,
_J_=6.2 Hz, 3H), 1.27–1.42 (m, 4H), 1.55 (d, _J_=6.9 Hz, 1H), 1.78–1.87 (m, 1H), 1.98–2.27 (m, 3H), 1.99 (s, 3H), 2.43–2.55 (m, 1H), 2.56–2.66 (m, 1H), 3.11 (s, 3H), 3.39–3.46 (m, 1H), 3.55
(dd, _J_=10.3, 3.2 Hz, 1H), 3.83 (br dd, _J_=3.2, 0.9 Hz, 1H), 3.86–3.97 (m, 1H), 4.10 (dd, _J_=10.3, 5.7 Hz, 1H), 4.33 (dq, _J_=6.9, 2.9 Hz, 1H), 4.42–4.48 (m, 1H), 4.52–4.59 (m, 1H), 4.61
(dd, _J_=9.9, 2.9 Hz, 1H), 5.25 (d, _J_=5.7 Hz, 1H) and 8.11 (s, 1H). 7(_S_)-1′-_N_-ACETYL-1′-DEMETHYL-7-DEOXY-7-[5-{5-(METHYLAMINO)THIAZOL-4-YL}-1,3,4-THIADIAZOL-2-YLTHIO]LINCOMYCIN (44) To
a solution of compound 35 (30.2 mg, 0.050 mmol) in MeOH (0.5 ml) at 0 °C was added acetic anhydride (7.0 μl, 0.074 mmol) and stirred at 0 °C for 1.5 h. The mixture was concentrated under
reduced pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=10/1/0.1) to obtain the title compound (10.8 mg, 33%) as an off-white solid. [α]D25 +56.5°
(_c_ 1.25, MeOH); ESI-MS _m/z_ 647 (M+H)+ as C25H38N6O6S4; TOF-ESI-HRMS (M+H)+ calcd. for C25H38N6O6S4: 647.1814, found: 647.1807; 1H NMR (400 MHz, CD3OD) δ 0.87–0.98 (m, 3H), 1.25–1.42 (m,
4H), 1.49 (d, _J_=7.0 Hz, 3H), 1.82–1.95 (m, 1H), 1.98 (s, 3H), 2.01–2.08 (m, 1H), 2.09 (s, 3H), 2.35–2.50 (m, 1H), 3.12 (s, 3H), 3.14–3.20 (m, 1H), 3.56 (dd, _J_=10.2, 3.3 Hz, 1H),
3.79–3.87 (m, 1H), 3.99 (br dd, _J_=3.3, 1.0 Hz, 1H), 4.09 (dd, _J_=10.2, 5.6 Hz, 1H), 4.27 (dq, _J_=7.0, 2.4 Hz, 1H), 4.36–4.43 (m, 1H), 4.48 (dd, _J_=8.8, 2.7 Hz, 1H), 4.61 (dd, _J_=10.0,
2.4 Hz, 1H) and 8.12 (s, 1H). 7(_S_)-1′-DEMETHYL-7-DEOXY-7-[5-{5-(METHYLAMINO)THIAZOL-4-YL}-1,3,4-THIADIAZOL-2-YLTHIO]-1′-_N_-(4-METHYLTHIAZOL-5-YLMETHYL)LINCOMYCIN (45) Compound 35 (24.0
mg, 0.04 mmol), 4-methylthiazole-5-carbaldehyde (16.2 mg, 0.13 mmol), AcOH (one drop) and NaBH(OAc)3 (25.5 mg, 0.12 mmol) in MeOH (0.5 ml) were treated at room temperature for 15 h according
to the similar procedure as described for the preparation of 38 to afford 45 (6.4 mg, 22.0%) as a colorless solid. [α]D26 +26° (_c_ 0.05, MeOH); ESI-MS _m/z_ 716 (M+H)+ as C28H41N7O5S5;
TOF-ESI-HRMS (M+H)+ calcd. for C28H41N7O5S5: 716.1851, found: 716.1854; 1H NMR (400 MHz, CD3OD) δ 0.75–0.86 (m, 3H), 1.15–1.26 (m, 4H), 1.42 (d, _J_=6.8 Hz, 3H), 1.68–1.78 (m, 1H), 1.89–2.15
(m, 3H), 1.94 (s, 3H), 2.27 (s, 3H), 3.00 (s, 3H), 3.09 (dd, _J_=8.2, 6.2 Hz, 1H), 3.24–3.31 (m, 1H), 3.50 (dd, _J_=10.2, 3.2 Hz, 1H), 3.72 (d, _J_=14.3 Hz, 1H), 3.79 (br dd, _J_=3.2, 1.0
Hz, 1H), 3.85 (d, _J_=14.3 Hz, 1H), 4.01 (dd, _J_=10.2, 5.5 Hz, 1H), 4.13–4.21 (m, 1H), 4.28–4.34 (m, 1H), 4.44 (dd, _J_=8.8, 4.6 Hz, 1H), 5.16 (d, _J_=5.5 Hz, 1H, 8.03 (s, 1H) and 8.69 (s,
1H). 1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-PROPYLLINCOMYCIN (46) Compound 17 (1.55 g, 5.19 mmol), 1-hydroxybenzotriazole (1.05 g, 7.78 mmol), _N_,_N_′-dicyclohexylcarbodiimide (1.61
g, 7.78 mmol) and MTL (1.97 g, 7.78 mmol) in DMF (15.0 ml) were treated for 14 h according to the similar procedure as described for the preparation of 18 to afford 46. The total amount of
this compound was used without purification to synthesize 47. For the qualified analytical purpose, the above crude 46 was purified by column chromatography (ethyl acetate only) to obtain
the title compound as a colorless solid. ESI-MS _m/z_ 535 (M+H)+ as C25H46N2O8S; 1H NMR (400 MHz, CD3OD) δ 0.76–0.90 (m, 6H), 1.04–1.32 (m, 11H), 1.35, 1.36 (s x 2, 9H), 1.57–1.72 (m, 1H),
1.97, 1.99 (s x 2, 3H), 2.00–2.15 (m, 1H), 3.03 (br t, _J_=10.4 Hz, 1H), 3.29–3.56 (m, 2H), 3.67–3.90 (m, 1H), 3.91–4.36 (m, 5H) and 5.15 (br d, _J_=5.4 Hz, 1H).
1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-PROPYL-2,3,4-TRIS-_O_-(TRIMETHYLSILYL)LINCOMYCIN (47) Compound 46 (crude), trimethylchlorosilane (3.32 ml, 25.9 mmol) and hexamethyldisilazane
(5.44 ml, 25.9 mmol) in pyridine (5.0 ml) were treated for 1 h according to the similar procedure as described for the preparation of 23 and then the crude compound and 6 n acetic acid (1.55
ml) in methanol (29.4 ml) were treated for 1 h according to the similar procedure as described for the preparation of 23 to afford 47 (1.66 g, 41.6% in three steps from 17) as a colorless
solid. ESI-MS _m/z_ 751 (M+H)+ as C34H70N2O8SSi3; 1H NMR (400 MHz, CDCl3) δ 0.14 (s, 18H), 0.18 (s, 9H), 0.82–0.98 (m, 6H), 1.08–1.35 (m, 11H), 1.46 (s, 9H), 1.80–2.35 (m, 1H), 2.06 (s, 3H),
2.66–3.30 (m, 2H), 3.36–4.41 (m, 8H) and 5.13–5.24 (m, 1H). 7(_S_)-1′-DEMETHYL-7-DEOXY-4′-_N_-PROPYL-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YLTHIO}LINCOMYCIN (48) To a solution of compound
47 (400 mg, 0.53 mmol) in THF (4 ml) at 0°C were added triphenylphosphine (209.6 mg, 0.80 mmol), diethylazodicarboxylate (0.15 ml, 0.80 mmol), 5-(2-nitrophenyl)-1,3,4-thiadiazole-2-thiol
(296.7 mg, 1.24 mmol) and stirred at room temperature for 6 h. The solution was purified by preparative TLC (hexane/ethyl acetate=2/1) to obtain
7(_S_)-1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-7-deoxy-4′-_n_-propyl-7-{5-(2-nitrophenyl)-1,3,4-thiadiazol-2-ylthio}-2,3,4-tris-_O_-(trimethylsilyl)lincomycin (235.4 mg with unseparable
impurity). To this crude compound (235.4 mg 0.24 mmol) was added 2,2,2-trifluoroacetic acid (1 ml) at 0 °C and stirred at room temperature for 30 min. The solution was concentrated under
reduced pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq NH4OH=9/2/0.2) to obtain the title compound (57.3 mg, 36.1% in 2 steps from 47) as a colorless
solid. [α]D26 +91.0° (_c_ 0.40, MeOH); ESI-MS _m/z_ 656 (M+H)+ as C28H41N5O7S3; TOF-ESI-HRMS (M+H)+ calcd. for C28H41N5O7S3: 656.2246, found: 656.2239; 1H NMR (400 MHz, CD3OD) δ 0.86–0.97
(m, 6H), 1.22–1.42 (m, 8H), 1.56 (d, _J_=6.8 Hz, 3H), 1.63 (dd, _J_=13.0, 8.2 Hz, 1H), 2.00 (s, 3H), 2.11–2.19 (m, 1H), 2.79–2.88 (m, 2H), 3.56 (dd, _J_=10.2, 3.2 Hz, 1H), 3.80–3.86 (m, 1H),
3.93 (t, _J_=8.4 Hz, 1H), 4.10 (dd, _J_=10.2, 5.6 Hz, 1H), 4.38–4.43 (m, 1H), 4.45 (dq, _J_=6.8, 2.7 Hz, 1H), 4.64 (dd, _J_=10.0, 2.7 Hz, 1H), 5.27 (d, _J_=5.6 Hz, 1H), 7.75–7.86 (m, 3H)
and 8.07–8.13 (m, 1H). 1′-_N_-(_TERT_-BUTOXYCARBONYL)-4′-{3-(_TERT_-BUTYLDIMETHYLSILYLOXY)PROPYL}-1′-DEMETHYL-4′-DEPROPYLLINCOMYCIN (49) Compound 11 (3.02, crude), 1-hydroxybenzotriazole
(1.35 g, 10.01 mmol), _N_,_N_′-dicyclohexylcarbodiimide (2.07 g, 10.01 mmol) and MTL (2.54 g, 10.01 mmol) in DMF (15.0 ml) were treated for 13 h according to the similar procedure as
described for the preparation of 19 to afford 49 (3.0 g, 62.5% in 3 steps from 8) as a colorless solid. ESI-MS _m/z_ 623 (M+H)+ as C28H54N2O9SSi; 1H NMR (400 MHz, CD3OD) δ 0.06 (s, 6H), 0.90
(s, 9H), 1.15–1.27 (m, 3H), 1.37–1.64 (m, 4H), 1.44, 1.47 (s x 2, 9H), 1.74–1.91 (m, 1H), 2.06, 2.07 (s x 2, 3H), 2.09–2.18 (m, 1H), 2.30–2.45 (m, 1H), 2.94 (br t, _J_=10.0 Hz, 1H),
3.53–3.72 (m, 4H), 3.74–3.93 (m, 1H), 3.98–4.17 (m, 3H), 4.24 (br dd, _J_=8.9, 1.3 Hz, 1H), 4.27–4.46 (m, 1H) and 5.24 (d, _J_=5.4 Hz, 1H).
1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-4′-DEPROPYL-4′-{03-(_TERT_-BUTYLDIMETHYLSILYLOXY)PROPYL}- 2,3,4-TRIS-_O_-(TRIMETHYLSILYL)LINCOMYCIN (50) Compound 49 (3.0 g, 4.82 mmol),
trimethylchlorosilane (3.08 ml, 24.1 mmol) and hexamethyldisilazane (5.05 ml, 24.1 mmol) in pyridine (10 ml) were treated for 20 min according to the similar procedure as described for the
preparation of 23 and then the crude compound and 6 n acetic acid (1.45 ml) in methanol (27 ml) were treated for 80 min according to the similar procedure as described for the preparation of
23 to afford 50 (3.02 g, 74.8% in two steps from 49) as a colorless solid. ESI-MS _m/z_ 839 (M+H)+ as C37H78N2O9SSi4; 1H NMR (400 MHz, CDCl3) δ 0.04 (s, 6H), 0.07–0.20 (m, 27H), 0.89 (s,
9H), 1.05–1.22 (m, 3H), 1.35–1.90 (m, 5H), 1.48 (s, 9H), 2.07 (s, 3H), 2.11–3.25 (m, 3H), 3.44–3.66 (m, 4H), 3.70–3.89 (m, 1H), 3.91–4.18 (m, 3H), 4.19–4.42 (m, 2H) and 5.19 (br d, _J_=5.6
Hz, 1H). 7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-4′-(3-HYDROXYPROPYL)-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YLTHIO}LINCOMYCIN (51) To a solution of compound
50 (2.87 g, 3.41 mmol) in THF (15 ml) at 0 °C were added triphenylphosphine (1.34 g, 5.12 mmol), diethylazodicarboxylate (932 μl, 5.12 mmol) and 5-(2-nitrophenyl)-1,3,4-thiadiazole-2-thiol
(1.26 g, 5.29 mmol) and stirred at room temperature for 10 h. The solution was substituted by toluene, purified by silica gel column chromatography (hexane to hexane/ethyl acetate=4/1) to
obtain
1′-_N_-(_tert_-butoxycarbonyl)-4′-{3-(_tert_-butyldimethylsilyloxy)propyl}-1′-demethyl-7-deoxy-4′-depropyl-7-{5-(2-nitrophenyl)-1,3,4-thiadiazol-2-ylthio}-2,3,4-tris-_O_-(trimethylsilyl)lincomycin
(2.62 g, crude). To this crude compound (2.62 g) was added 1 m THF solution of _tetra_-_n_-butyl ammonium fluoride (14.8 ml, 14.8 mmol) and acetic acid (0.848 ml, 14.8 mmol) and stirred at
room temperature for 5 h. The mixture was diluted with brine and ethyl acetate, extracted with ethyl acetate, and then the organic phase was dried over Na2SO4, filtrated and concentrated
under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=1/1 to ethyl acetate only to ethyl acetate/MeOH=10/1) to obtain the title
compound (1.74 g with unseparable impurity (96% in two steps as reference yield)). FAB-MS _m/z_ 752 (M+Na)+ as C30H43N5O10S3.
7(_S_)-4′-(3-AMINOPROPYL)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YLTHIO}LINCOMYCIN (52) To a solution of compound 51 (500 mg,
0.685 mmol), sodium azide (223 mg, 3.43 mmol) and triphenylphosphine (359.3 mg, 1.37 mmol) in DMF (7 ml) was added tetrabromomethane (454.3 mg, 1.37 mmol) and stirred at 50 °C for 2 h. The
mixture was diluted with brine and ethyl acetate, extracted with ethyl acetate and then the organic phase was dried over Na2SO4, filtrated and concentrated under reduced pressure. The
resulting residue was purified by silica gel column chromatography (chloroform/MeOH=10/1) to obtain
7(_S_)-4′-(3-azidopropyl)-1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-7-deoxy-4′-depropyl-7-{5-(2-nitrophenyl)-1,3,4-thiadiazol-2-ylthio}lincomycin (468 mg with unseparable impurity (91% as
reference yield)). To a solution of this crude compound (239 mg, 0.317 mmol) in THF (3 ml) was added triphenylphosphine (249.0 mg, 0.95 mmol), stirred at room temperature for 1 h, and then
H2O was added and stirred at 50 °C for 2 h. The mixture was diluted with brine, ethyl acetate and 1 n HCl (400 μl), washed with ethyl acetate and then the aqueous phase was added to the
saturated aqueous NaHCO3, extracted with CHCl3, dried over Na2SO4, filtrated and concentrated under reduced pressure to obtain the title compound (229 mg with unseparable impurity (99% as
reference yield)). 7(_S_)-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-4′-(3-DIMETHYLAMINOPROPYL)-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YLTHIO}LINCOMYCIN (53) Compound 52 (69.0 mg, 0.093 mmol), 36%
aqueous formaldehyde (23.1 μl, 0.28 mmol), AcOH (79.5 μl, 1.39 mmol) and NaBH(OAc)3 (294.3 mg, 1.39 mmol) in MeOH (1 ml) were treated at room temperature for 20 min according to the similar
procedure as described for the preparation of 38 to afford
7(_S_)-1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-7-deoxy-4′-depropyl-4′-(3-dimethylaminopropyl)-7-{5-(2-nitrophenyl)-1,3,4-thiadiazol-2-ylthio}lincomycin (75.0 mg, crude). To this crude
compound (75.0 mg) was added 2,2,2-trifluoroacetic acid (1 ml) at 0 °C and stirred at room temperature for 30 min. The solution was concentrated under reduced pressure. The resulting residue
was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/4/0.4) to obtain the title compound (35.0 mg, 57.6% in two steps from 52) as a colorless solid. [α]D25 +83.7° (_c_ 0.45, MeOH);
ESI-MS m/z 657 (M+H)+ as C27H40N6O7S3; TOF-ESI-HRMS (M+H)+ calcd. for C27H40N6O7S3: 657.2199, found: 657.2193; 1H NMR (400 MHz, CD3OD) δ 1.34–1.44 (m, 2H), 1.48–1.64 (m, 2H), 1.57 (d,
_J_=7.0 Hz, 3H), 1.76–1.88 (m, 1H), 2.01 (s, 3H), 2.04–2.13 (m, 2H), 2.34 (s, 6 H), 2.46 (t, _J_=7.8 Hz, 2H), 2.56 (dd, _J_=10.3, 7.8 Hz, 1H), 3.24 (dd, _J_=10.3, 6.8 Hz, 1H), 3.56 (dd,
_J_=10.2, 3.3 Hz, 1H), 3.81 (dd, _J_=9.3, 3.9 Hz, 1H), 3.83 (br dd, _J_=3.3, 0.8 Hz, 1H), 4.10 (dd, _J_=10.2, 5.6 Hz, 1H), 4.41 (br dd, _J_=9.9, 0.8 Hz, 1H), 4.46 (dq, _J_=7.0, 2.8 Hz, 1H),
4.62 (dd, _J_=9.9, 2.8 Hz, 1H), 5.28 (d, _J_=5.6 Hz, 1H), 7.75–7.87 (m, 3H) and 8.06–8.15 (m, 1H).
7(_S_)-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-4′-(3-METHOXYPROPYL)-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YLTHIO}LINCOMYCIN (54) Compound 24 (266.0 mg, 0.36 mmol), triphenylphosphine (142.0 mg,
0.54 mmol), diethylazodicarboxylate (98 μl, 0.54 mmol) and 5-(2-nitrophenyl)-1,3,4-thiadiazole-2-thiol (132.0 mg, 0.56 mmol) in THF (2 ml) were treated for 18 h according to the similar
procedure as described for the preparation of 31 to afford
7(_S_)-1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-7-deoxy-4′-depropyl-4′-(3-methoxypropyl)-7-{5-(2-nitrophenyl)-1,3,4-thiadiazol-2-ylthio}-2,3,4-tris-_O_-(trimethylsilyl)lincomycin (325.0 mg
as crude). To this crude compound (325.0 mg) was added 2,2,2-trifluoroacetic acid (1 ml) at 0 °C and stirred at room temperature for 15 min. The solution was concentrated under reduced
pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound (112.0 mg, 48.3% in two steps from 24) as a colorless solid.
[α]D24 +82.6° (_c_ 0.45, MeOH); ESI-MS _m/z_ 644 (M+H)+ as C26H37N5O8S3; TOF-ESI-HRMS (M+H)+ calcd. for C26H37N5O8S3: 644.1883, found: 644.1880; 1H NMR (400 MHz,CD3OD) δ 1.40–1.52 (m, 2H),
1.54–1.65 (m, 2H), 1.57 (d, _J_=6.9 Hz, 3H), 1.89–1.97 (m, 1H), 2.01 (s, 3H), 2.09–2.23 (m, 2H), 2.68 (dd, _J_=10.7, 8.0 Hz, 1H), 3.30 (s, 3H), 3.32–3.36 (m, 1H), 3.39 (t, _J_=6.2, 2H), 3.57
(dd, _J_=10.2, 3.2 Hz, 1H), 3.85 (br dd, _J_=3.2, 0.8 Hz, 1H), 4.02 (dd, _J_=9.1, 4.1 Hz, 1H), 4.11 (dd, _J_=10.2, 5.6 Hz, 1H), 4.42 (br dd, _J_=10.0, 0.8 Hz, 1H), 4.47 (dq, _J_=6.9, 2.7
Hz, 1H), 4.66 (dd, _J_=10.0, 2.7 Hz, 1H), 5.28 (d, _J_=5.6 Hz, 1H), 7.76–7.87 (m, 3H) and 8.06–8.14 (m, 1H).
7(_S_)-4′-_I_-BUTYL-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-7-(5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YL)THIOLINCOMYCIN (55) Compound 25 (204.0 mg, 0.28 mmol), triphenylphosphine (109.1 mg, 0.42
mmol), diethylazodicarboxylate (75.8 μl, 0.42 mmol) and 5-(2-nitrophenyl)-1,3,4-thiadiazole-2-thiol (103.0 mg, 0.43 mmol) in THF (2 ml) were treated for 14.5 h according to the similar
procedure as described for the preparation of 31 to afford
7(_S_)-1′-_N_-(_tert_-butoxycarbonyl)-4′-_i_-butyl-1′-demethyl-7-deoxy-4′-depropyl-7-{5-(2-nitrophenyl)-1,3,4-thiadiazol-2-ylthio}-2,3,4-tris-_O_-(trimethylsilyl)lincomycin (170.0 mg as
crude). To this crude compound (170.0 mg) was added 2,2,2-trifluoroacetic acid (1 ml) at 0 °C and stirred at room temperature for 30 min. The solution was concentrated under reduced
pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound (39.0 mg, 22.5% in two steps from 25) as a colorless solid.
[α]D24 +85.8° (_c_ 0.54, MeOH); ESI-MS _m/z_ 628 (M+H)+ as C26H37N5O7S3; TOF-ESI-HRMS (M+H)+ calcd. for C26H37N5O7S3: 628.1933, found: 628.1936; 1H NMR (400 MHz, CD3OD) δ 0.90 (d, _J_=5.3
Hz, 3H), 0.91 (d, _J_=5.4 Hz, 3H), 1.23–1.34 (m, 2H), 1.53–1.64 (m, 1H), 1.57 (d, _J_=6.8 Hz, 3H), 1.81–1.92 (m, 1H), 2.00 (s, 3H), 2.04–2.15 (m, 1H), 2.15–2.27 (m, 1H), 2.58 (dd, _J_=10.4,
8.7 Hz, 1H), 3.26–3.30 (m, 1H), 3.56 (dd, _J_=10.2, 3.2 Hz, 1H), 3.84 (br dd, _J_=3.2, 0.8 Hz, 1H), 3.92 (dd, _J_=9.2, 4.3 Hz, 1H), 4.10 (dd, _J_=10.2, 5.6 Hz, 1H), 4.41 (br dd, _J_=10.0,
0.8 Hz, 1H), 4.47 (dq, _J_=6.8, 2.8 Hz, 1H), 4.65 (dd, _J_=10.0, 2.8 Hz, 1H), 5.28 (d, _J_=5.6 Hz, 1H), 7.76–7.87 (m, 3H) and 8.08–8.13 (m, 1H).
7(_S_)-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YLTHIO}-4′-_N_-PENTYLLINCOMYCIN (56) Compound 26 (400 mg, 0.54 mmol), triphenylphosphine (213.5 mg, 0.81 mmol),
diethylazodicarboxylate (150 μl, 0.81 mmol) and 5-(2-nitrophenyl)-1,3,4-thiadiazole-2-thiol (201.4 mg, 0.84 mmol) in THF (4 ml) were treated for 24 h according to the similar procedure as
described for the preparation of 31 to afford
7(_S_)-1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-7-deoxy-4′-depropyl-7-{5-(2-nitrophenyl)-1,3,4-thiadiazol-2-ylthio}-4′-_n_-pentyl-2,3,4-tris-_O_-(trimethylsilyl)lincomycin (374.0 mg as
crude). To this crude compound (374 mg) was added 2,2,2-trifluoroacetic acid (1 ml) at 0 °C and stirred at room temperature for 30 min. The solution was concentrated under reduced pressure.
The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound (72.6 mg, 20.9% in two steps from 26) as a colorless solid. [α]D25
+34.3° (_c_ 0.10, MeOH); ESI-MS _m/z_ 642 (M+H)+ as C27H39N5O7S3; TOF-ESI-HRMS (M+H)+ calcd. for C27H39N5O7S3: 642.2090, found: 642.2083; 1H NMR (400 MHz, CD3OD) δ 0.80–0.95 (m, 3H),
1.21–1.43 (m, 8H), 1.57 (d, _J_=7.0 Hz, 3H), 1.81–1.95 (m, 1H), 2.02–2.14 (m, 2H), 2.09 (s, 3H), 2.62 (dd, _J_=10.4, 7.9 Hz, 1H), 3.27 (dd, _J_=10.5, 6.8 Hz, 1H), 3.61 (dd, _J_=10.2, 3.2 Hz,
1H), 3.83 (br dd, _J_=3.2, 0.9 Hz, 1H), 3.91 (dd, _J_=9.1, 4.2 Hz, 1H), 4.14 (dd, _J_=10.2, 5.6 Hz, 1H), 4.34–4.40 (m, 1H), 4.42 (dq, _J_=7.0, 2.9 Hz, 1H), 4.57 (dd, _J_=10.0, 2.9 Hz, 1H),
5.31 (d, _J_=5.6 Hz, 1H), 7.71–7.87 (m, 3H) and 8.07–8.14 (m, 1H). 7(_S_)-7-DEOXY-4′-DEPROPYL-7-{5-(2-NITROPHENYL)-1,3,4-THIADIAZOL-2-YLTHIO}-4′-_N_-PENTYLLINCOMYCIN (57) Compound 56 (56.1
mg, 0.087 mmol), 36% aqueous formaldehyde (22 μl, 0.26 mmol), AcOH (30 μl, 0.52 mmol) and NaBH(OAc)3 (55.5 mg, 0.26 mmol) in MeOH (0.6 ml) were treated at room temperature for 1 h according
to the similar procedure as described for the preparation of 38 to afford 57 (49.1 mg, 85.7%) as a colorless solid. [α]D25 +91.6° (_c_ 1.16, MeOH); ESI-MS _m/z_ 656 (M+H)+ as C28H41N5O7S3;
TOF-ESI-HRMS (M+H)+ calcd. for C28H41N5O7S3: 656.2246, found: 656.2238; 1H NMR (400 MHz, CD3OD) δ 0.83–0.92 (m, 3H), 1.21–1.42 (m, 8H), 1.57 (d, _J_=7.0 Hz, 3H), 1.78–1.91 (m, 1H), 1.96–2.10
(m, 2H), 2.01 (s, 3H), 2.12–2.27 (m, 1H), 2.39 (s, 3H), 3.02 (dd, _J_=10.5, 5.1 Hz, 1H), 3.23–3.29 (m, 1H), 3.57 (dd, _J_=10.2, 3.2 Hz, 1H), 3.81 (dd, _J_=3.2, 0.7 Hz, 1H), 4.11 (dd,
_J_=10.2, 5.6 Hz, 1H), 4.39–4.49 (m, 1H), 4.60 (dd, _J_=9.8, 3.2 Hz, 1H), 5.27 (d, _J_=5.6, 1H), 7.75–7.85 (m, 3H) and 8.06–8.11 (m, 1H).
7(_S_)-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-7-[5-{5-(METHYLAMINO)THIAZOL-4-YL}-1,3,4-THIADIAZOL-2-YLTHIO]-4′-_N_-PENTYLLINCOMYCIN (58) Compound 26 (400 mg, 0.54 mmol), triphenylphosphine (213.5
mg, 0.81 mmol), diethylazodicarboxylate (150 μl, 0.81 mmol) and 5-{5-(methylamino)thiazol-4-yl}-1,3,4-thiadiazole-2-thiol (193.9 mg, 0.84 mmol) in THF (4 ml) were treated for 24 h according
to the similar procedure as described for the preparation of 31 to afford
7(_S_)-1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-7-deoxy-4′-depropyl-7-[5-{5-(methylamino)thiazol-4-yl}-1,3,4-thiadiazol-2-ylthio]-4′-_n_-pentyl-2,3,4-tris-_O_-(trimethylsilyl)lincomycin
(109.8 mg, 21.3%). To the compound (109.8 mg, 0.12 mmol) was added 2,2,2-trifluoroacetic acid (1 ml) at 0 °C and stirred at room temperature for 30 min. The solution was concentrated under
reduced pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound (59.0 mg, 80.6%) as a colorless solid. [α]D25 +48.5°
(_c_ 0.22, MeOH); ESI-MS _m/z_ 633 (M+H)+ as C25H40N6O5S4; TOF-ESI-HRMS (M+H)+ calcd. for C25H40N6O5S4: 633.2021, found: 633.2021; 1H NMR (400 MHz, CD3OD) δ 0.83–0.94 (m, 3H), 1.21–1.43 (m,
8H), 1.50 (d, _J_=6.8 Hz, 3H), 1.80–1.91 (m, 1H), 2.02 (s, 3H), 2.02–2.13 (m, 2H), 2.59 (dd, _J_=10.5, 8.0 Hz, 1H), 3.12 (s, 3H), 3.24–3.30 (m, 1H), 3.58 (dd, _J_=10.2, 3.2 Hz, 1H), 3.85 (br
dd, _J_=3.2, 0.7 Hz, 1H), 3.93 (dd, _J_=9.1, 4.0 Hz, 1H), 4.11 (dd, _J_=10.2, 5.6 Hz, 1H), 4.29 (dq, _J_=6.8, 2.7 Hz, 1H), 4.37–4.44 (m, 1H), 4.61 (dd, _J_=10.0, 2.7 Hz, 1H), 5.28 (d,
_J_=5.6 Hz, 1H) and 8.13 (s, 1H). 7(_S_)-7-DEOXY-4′-DEPROPYL-7-[5-{5-(METHYLAMINO)THIAZOL-4-YL}-1,3,4-THIADIAZOL-2-YLTHIO]-4′-_N_-PENTYLLINCOMYCIN (59) Compound 58 (35.8 mg, 0.057 mmol), 36%
aqueous formaldehyde (14 μl, 0.17 mmol), AcOH (20 μl, 0.34 mmol) and NaBH(OAc)3 (36.0 mg, 0.17 mmol) in MeOH (0.5 ml) were treated at room temperature for 1 h according to the similar
procedure as described for the preparation of 38 to afford 59 (29.4 mg, 80.3%) as an off-white solid. [α]D25 +64.5° (_c_ 0.30, MeOH); ESI-MS _m/z_ 647 (M+H)+ as C26H42N6O5S4; TOF-ESI-HRMS
(M+H)+ calcd. for C26H42N6O5S4: 647.2178, found: 647.2176; 1H NMR (400 MHz, CD3OD) δ 0.84–0.93 (m, 3H), 1.19–1.39 (m, 8H), 1.54 (d, _J_=6.9 Hz, 3H), 1.80–1.92 (m, 1H), 2.02 (s, 3H),
1.96–2.21 (m, 3H), 2.40 (s, 3H), 3.06–3.14 (m, 1H), 3.12 (s, 3H), 3.21 (dd, _J_=7.9, 5.5 Hz, 1H), 3.61 (dd, _J_=10.2, 3.1 Hz, 1H), 3.84 (dd, _J_=3.1, 0.6 Hz, 1H), 4.12 (dd, _J_=10.2, 5.6 Hz,
1H), 4.28 (dq, _J_=6.9, 2.9 Hz, 1H), 4.43–4.44 (m, 1H), 4.59 (dd, _J_=10.0, 2.9 Hz, 1H), 5.28 (d, _J_=5.6 Hz, 1H) and 8.14 (s, 1H).
7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-7-{4-(METHOXYCARBONYL)PHENYLTHIO}LINCOMYCIN (60) To a solution of compound 23 (5.0 g, 7.05 mmol) in CHCl3 (22 ml) were added Et3N
(2.45 ml, 17.6 mmol), methanesulfonyl chloride (1.1 ml, 14.1 mmol) and stirred at room temperature for 30 min. The mixture was added to saturated aqueous NaHCO3, extracted with CHCl3, dried
over Na2SO4 and concentrated under reduced pressure. To a solution of crude compound in DMF (50 ml) were added K2CO3 (2.92 g, 21.2 mmol) and methyl 4-mercaptobenzoate (2.37 g, 14.1 mmol),
stirred at 100 °C for 6 h and concentrated under reduced pressure. The resulting residue in MeOH (50 ml) was added to 1 n HCl (100 ml) and stirred at room temperature for 20 min. The mixture
was added to the saturated aqueous NaHCO3, then extracted with ethyl acetate, dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by
silica gel column chromatography (CHCl3/MeOH/28% aq. NH4OH=20/1/0.1) to obtain the title compound as a colorless solid (1.25 g, 27.6% in three steps from 23). ESI-MS (_m/z_) 643 (M+H)+ as
C30H46N2O9S2; 1H NMR (400 MHz, CD3OD) δ 0.86–1.00 (m, 3H), 1.25–1.65 (m, 16H), 1.45, 1.48 (s x 2, 9H), 1.73–1.99 (m, 1H), 1.79, 1.85 (s x 2, 3H), 2.07–2.20 (m, 1H), 2.22–2.42 (m, 1H),
2.92–3.01 (m, 1H), 3.53–3.61 (m, 1H), 3.63–3.74 (m, 1H), 3.88 (s, 3H), 3.90–4.15 (m, 3H), 4.28–4.50 (m, 2H), 4.54–4.68 (m, 1H), 5.24 (d, _J_=5.4 Hz, 1H), 7.39–7.46 (m, 2H) and 7.89–7.96 (m,
2H). 7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-7-(4-(METHOXYCARBONYL)PHENYL)THIO-4′-{(_E_)-PENT-2-ENYL}LINCOMYCIN (61) Compound 27 (1.45 g, 1.97 mmol), Et3N (0.69
ml, 4.93 mmol) and methanesulfonyl chloride (0.31 ml, 3.93 mmol) in CHCl3 (6.1 ml) were treated at room temperature for 30 min according to the similar procedure as described for the
preparation of 60 to afford 1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-4′-depropyl-7-_O_-methanesulfonyl-4′-{(_E_)-pent-2-enyl}-2,3,4-tris-_O_-(trimethylsilyl)lincomycin. To a solution of
crude compound were added K2CO3 (1.23 g, 8.87 mmol) and methyl 4-mercaptobenzoate (0.99 g, 5.91 mmol) in DMF (13.8 ml) and treated at 80 °C for 1 h. The resulting residue and 1 n HCl (14
ml) in MeOH (14 ml) were treated at room temperature for 20 min according to the similar procedure as described for the preparation of 60 to afford 61 (1.21 g, 92% in three steps from 27) as
a colorless solid. EI-MS _m/z_ 668 (M)+ as C32H48N2O9S2; 1H NMR (400 MHz, CD3OD) δ 0.95 (t, _J_=7.4 Hz, 3H), 1.33–1.44 (m, 3H), 1.45, 1.48 (s x 2, 9H), 1.80, 1.85 (s x 2, 3H), 1.87–2.17 (m,
6H), 2.26–2.40 (m, 1H), 3.01–3.09 (m, 1H), 3.51–3.67 (m, 2H), 3.88 (s, 3H), 3.91–4.12 (m, 3H), 4.27–4.46 (m, 2H), 4.53–4.64 (m, 1H), 5.24 (d, _J_=5.6 Hz, 1H), 5.31–5.45 (m, 1H), 5.47–5.59
(m, 1H), 7.36–7.44 (m, 2H) and 7.86–7.95 (m, 2H). 7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-7-{4-(METHOXYCARBONYL)PHENYLTHIO}-4′-_N_-PENTYLLINCOMYCIN (62)
Compound 61 (105 mg, 0.16 mmol) and Pd/C (100 mg) in MeOH (2 ml) were treated for 15 h according to the similar procedure as described for the preparation of 7 to afford 62 (97 mg, 92.1%) as
a colorless solid. ESI-MS _m/z_ 671 (M+H)+ as C32H50N2O9S2; 1H NMR (400 MHz, CD3OD) δ 0.18–0.92 (m, 3H), 1.15–1.52 (m, 11H), 1.44, 1.47 (s x 2, 9H), 1.71–1.93 (m, 1H), 1.78, 1.83 (s x 2,
3H), 2.05–2.18 (m, 1H), 2.20–2.36 (m, 1H), 2.95 (br t, _J_=10.0 Hz, 1H), 3.52–3.61 (m, 1H), 3.61–3.73 (m, 1H), 3.87 (s, 3H), 3.91–4.03 (m, 2H), 4.03–4.15 (m, 1H), 4.28–4.47 (m, 2H),
4.53–4.65 (m, 1H), 5.25 (d, _J_=5.6 Hz, 1H), 7.35–7.47 (m, 2H) and 7.85–7.95 (m, 2H). 7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-7-{4-(CARBOXYL)PHENYLTHIO}-1′-DEMETHYL-7-DEOXYLINCOMYCIN (63) To a
solution of compound 60 (761 mg, 1.18 mmol) in MeOH (20 ml) was added 1 n NaOH (1.78 ml, 1.78 mmol) and stirred at room temperature for 7 days. The mixture was added 1 n HCl (pH=3),
extracted with CHCl3, dried over Na2SO4 and concentrated under reduced pressure to obtain the title compound (705 mg, 94.7%) as an off-white solid. ESI-MS _m/z_ 629 (M+H)+ as C29H44N2O9S2.
7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-7-{4-(CARBOXYL)PHENYLTHIO}-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-4′-_N_-PENTYLLINCOMYCIN (64) Compound 62 (97 mg, 0.15 mmol) and 1 n NaOH (0.55 ml) in MeOH
(1.1 ml) were treated for 18 h according to the similar procedure as described for the preparation of 63 to afford 64 (91.3 mg, 96.1%) as a colorless solid. FAB-MS _m/z_ 695 (M+K)+ as
C31H48N2O9S2; 1H NMR (400 MHz, CD3OD) δ 0.82–0.92 (m, 3H), 1.20–1.45 (m, 11H), 1.46, 1.48 (s x 2, 9H), 1.75–1.95 (m, 1H), 1.81, 1.86 (s x 2, 3H), 2.09–2.19 (m, 1H), 2.21–2.37 (m, 1H),
2.90–3.02 (m, 1H), 3.54–3.62 (m, 1H), 3.63–3.75 (m, 1H), 3.90–4.05 (m, 2H), 4.05–4.15 (m, 1H), 4.30–4.49 (m, 2H), 4.53–4.67 (m, 1H), 5.27 (d, _J_=5.6 Hz, 1H), 7.37–7.45 (m, 2H) and 7.89–7.97
(m, 2H). 7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-7-{4-(MORPHOLINOCARBONYL)PHENYLTHIO}LINCOMYCIN (65) To a solution of compound 63 (200 mg, 0.32 mmol),
1-hydroxybenzotriazole (64.5 mg, 0.48 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide HCl salt (91.5 mg, 0.48 mmol) in DMF (2 ml) was added to morpholine (42 μl, 0.48 mmol) and
stirred at room temperature for 22 h. The mixture was added to saturated aqueous NaHCO3, extracted with ethyl acetate, dried over Na2SO4 and concentrated under reduced pressure. The
resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound (215.0 mg, 96.8%) as a colorless solid. ESI-MS _m/z_ 698 (M+H)+ as
C33H51N3O9S2; 1H NMR (400 MHz, CD3OD) δ 0.86–0.97 (m, 3H), 1.25–1.45 (m, 7H), 1.46, 1.48 (s x 2, 9H), 1.78–1.95 (m, 1H), 1.88, 1.91 (s x 2, 3H), 2.06–2.18 (m, 1H), 2.24–2.40 (m, 1H),
2.92–2.99 (m, 1H), 3.37–3.85 (m, 10H), 3.88–3.99 (m, 2H), 4.05–4.14 (m, 1H), 4.30–4.45 (m, 2H), 4.52–4.63 (m, 1H), 5.26 (d, _J_=5.6 Hz, 1H), 7.35–7.42 (m, 2H) and 7.43–7.48 (m, 2H).
7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-7-{4-(MORPHOLINOCARBONYL)PHENYLTHIO}-4′-_N_-PENTYLLINCOMYCIN (66) Compound 64 (91.3 mg, 0.14 mmol),
1-hydroxybenzotriazole (28.1 mg, 0.21 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide HCl salt (40.0 mg, 0.21 mmol) and morpholine (18 μl, 0.21 mmol) in DMF (1 ml) were treated for
62 h according to the similar procedure as described for the preparation of 65 to afford 66 (82.0 mg, 81.3%) as a colorless solid. FAB-MS _m/z_ 726 (M+H)+ as C35H55N3O9S2; 1H NMR (400 MHz,
CD3OD) δ 0.82–0.94 (m, 3H), 1.21–1.42 (m, 11H), 1.46, 1.48 (s x 2, 9H), 1.77–1.95 (m, 1H), 1.87, 1.91 (s x 2, 3H), 2.08–2.18 (m, 1H), 2.20–2.38 (m, 1H), 2.90–3.01 (m, 1H), 3.38–3.84 (m,
10H), 3.87–4.01 (m, 2H), 4.05–4.14 (m, 1H), 4.29–4.46 (m, 2H), 4.52–4.63 (m, 1H), 5.26 (d, _J_=5.6 Hz, 1H), 7.35–7.42 (m, 2H) and 7.42–7.48 (m, 2H).
7(_S_)-1′-DEMETHYL-7-DEOXY-7-{4-(MORPHOLINOCARBONYL)PHENYLTHIO}LINCOMYCIN (67) To the compound 65 (215 mg, 0.32 mmol) was added 2,2,2-trifluoroacetic acid (2 ml) at 0 °C and stirred for 40
min. The solution was concentrated under reduced pressure. The resulting residue was purified by preparative TLC (CHCl3/MeOH/28% aq. NH4OH=9/2/0.2) to obtain the title compound (104.0 mg,
56.5%) as a colorless solid. [α]D25 +70.5° (_c_ 0.42, MeOH); ESI-MS _m/z_ 598 (M+H)+ as C28H43N3O7S2; TOF-ESI-HRMS (M+H)+ calcd. for C28H43N3O7S2: 598.2621, found: 598.2623; 1H NMR (400 MHz,
CD3OD) δ 0.88–0.97 (m, 3H), 1.30–1.44 (m, 4H), 1.33 (d, _J_=7.0 Hz, 3H), 1.80–1.93 (m, 1H), 1.90 (s, 3H), 2.05–2.18 (m, 2H), 2.65 (dd, _J_=10.4, 8.2 Hz, 1H), 3.26–3.32 (m, 1H), 3.35–3.56
(m, 2H), 3.56 (dd, _J_=10.2, 3.3 Hz, 1H), 3.52–3.85 (m, 6H), 3.79 (br dd, _J_=3.3, 0.8 Hz, 1), 3.90–3.98 (m, 2H), 4.08 (dd, _J_=10.2, 5.6 Hz, 1H), 4.37 (br dd, _J_=9.9, 0.8 Hz, 1H), 4.54
(dd, _J_=9.9, 2.7 Hz, 1H), 5.25 (d, _J_=5.6 Hz, 1H), 7.35–7.41 (m, 2H) and 7.43–7.49 (m, 2H).
7(_S_)-1′-DEMETHYL-7-DEOXY-4′-DEPROPYL-7-{4-(MORPHOLINOCARBONYL)PHENYLTHIO}-4′-_N_-PENTYLLINCOMYCIN (68) Compound 66 (82.0 mg, 0.11 mmol) and 2,2,2-trifluoroacetic acid (1 ml) were treated
at −15 to 0 °C for 40 min according to the similar procedure as described for the preparation of 67 to afford 68 (57.0 mg, 80.6%) as a colorless solid. [α]D26 +74.0° (_c_ 0.51, MeOH); ESI-MS
_m/z_ 626 (M+H)+ as C30H47N3O7S2; TOF-ESI-HRMS (M+H)+ calcd. for C30H47N3O7S2: 626.2934, found: 626.2924; 1H NMR (400 MHz, CD3OD) δ 0.83–0.93 (m, 3H), 1.23–1.44 (m, 8H), 1.33 (d, _J_=6.9
Hz, 3H), 1.77–1.88 (m, 1H), 1.90 (s, 3H), 1.99–2.15 (m, 2H), 2.61 (dd, _J_=10.4, 8.2 Hz, 1H), 3.24 (dd, _J_=10.4, 7.0 Hz, 1H), 3.36–3.57 (m, 2H), 3.56 (dd, _J_=10.2, 3.2 Hz, 1H), 3.57–3.84
(m, 6H), 3.78 (br dd, _J_=3.2, 0.7 Hz, 1H), 3.88 (dd, _J_=9.5, 4.0 Hz, 1H), 3.95 (dq, _J_=6.9, 2.6 Hz, 1H), 4.08 (dd, _J_=10.2, 5.6 Hz, 1H), 4.36 (br dd, _J_=9.9, 0.7 Hz, 1H), 4.53 (dd,
_J_=9.9, 2.6 Hz, 1H), 5.25 (d, _J_=5.6 Hz, 1H), 7.34–7.41 (m, 2H) and 7.42–7.50 (m, 2H). 7(_S_)-7-DEOXY-4′-DEPROPYL-7-{4-(MORPHOLINOCARBONYL)PHENYLTHIO}-4′-_N_-PENTYLLINCOMYCIN (69) Compound
68 (31.0 mg, 0.050 mmol), 36% aqueous formaldehyde (12 μl, 0.15 mmol), AcOH (17 μl, 0.30 mmol) and NaBH(OAc)3 (31.6 mg, 0.15 mmol) in MeOH (0.5 ml) were treated at room temperature for 30
min according to the similar procedure as described for the preparation of 38 to afford 69 (31.0 mg, 97.8%) as a colorless solid. [α]D26 +68° (_c_ 0.12, MeOH); ESI-MS _m/z_ 640 (M+H)+ as
C31H49N3O7S2; TOF-ESI-HRMS (M+H)+ calcd. for C31H49N3O7S2: 640.3090, found: 640.3080; 1H NMR (400 MHz, CD3OD) δ 0.85–0.94 (m, 3H), 1.23–1.43 (m, 8H), 1.35 (d, _J_=6.8 Hz, 3H), 1.81–1.92 (m,
1H), 1.91 (s, 3H), 2.03 (ddd, _J_=13.0, 7.8, 5.0 Hz, 1H), 2.08–2.25 (m, 2H), 2.44 (s, 3H), 3.06 (dd, _J_=10.5, 4.9 Hz, 1H), 3.25–3.30 (m, 1H), 3.38–3.84 (m, 9H), 3.58 (dd, _J_=10.2, 3.2 Hz,
1H), 3.97 (dq, _J_=6.8, 2.6 Hz, 1H), 4.10 (dd, _J_=10.2, 5.6 Hz, 1H), 4.33–4.39 (m, 1H), 4.51 (dd, _J_=9.7, 2.6 Hz, 1H), 5.26 (d, _J_=5.6 Hz, 1H), 7.37–7.42 (m, 2H) and 7.45–7.50 (m, 2H).
7(_S_)-7-ACETYLTHIO-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXYLINCOMYCIN (70) To a solution of compound 23 (500.3 mg, 0.71 mmol) in CH2Cl2 (10 ml) were added Et3N (0.25 ml, 1.77
mmol) and methanesulfonyl chloride (0.11 ml, 1.39 mmol), stirred at 0 °C for 1 h. The mixture was added to saturated aqueous NH4Cl, extracted with ethyl acetate, washed with 25% brine, dried
over Na2SO4 and concentrated under reduced pressure. To a solution of this crude compound in DMF (8 ml) was added AcSK (501.7 mg, 4.39 mmol), stirred at 60°C for 10 h. The mixture was added
to the saturated aqueous NaHCO3, then extracted with ethyl acetate, dried over Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel
column chromatography (hexane/ethyl acetate=10/1 to 2/1) to obtain 7(_S_)-7-acetylthio-1′-_N_-(_tert_-butoxycarbonyl)-1′-demethyl-7-deoxy-2,3,4-tris-_O_-(trimethylsilyl)lincomycin as a
colorless solid (217.8 mg, 40.2% in two steps from 23). To a solution of this intermediate in MeOH (2.2 ml) was added 1 n HCl (2.2 ml) and stirred at room temperature for 1 h. The mixture
was added to saturated aqueous NaHCO3, extracted with ethyl acetate, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue was purified by silica gel column
chromatography (chloroform/MeOH=40/1) to obtain the title compound (137 mg, 87.6%) as a colorless solid. ESI-MS _m/z_ 551 (M+H)+ as C24H42N2O8S2; 1H NMR (400 MHz, CD3OD) δ 0.86–0.98 (m, 3H),
1.26–1.41 (m, 7H), 1.46 (s, 9H), 1.76–1.96 (m, 1H), 2.01, 2.03 (s, 3H), 1.95–2.17 (m, 1H), 2.20–2.38 (m, 1H), 2.32 (s, 3H), 2.95 (t, _J_=9.8 Hz, 1H), 3.51 (dd, _J_=10.2, 3.3 Hz, 1H), 3.66
(dd, _J_=10.0, 7.6 Hz, 1H), 3.81–4.01 (m, 2H), 4.01–4.12 (m, 1H), 4.13–4.37 (m, 2H), 4.41–4.50 (m, 1H) and 5.21 (d, _J_=5.5 Hz, 1H).
7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-7-MERCAPTOLINCOMYCIN (71) To a solution of compound 70 (137 mg, 0.25 mmol) in MeOH (1.5 ml) was added sodium methoxide (43.2 mg,
0.76 mmol) and stirred at room temperature for 1.5 h. The mixture was diluted with 8% aqueous NaHCO3, extracted with ethyl acetate, dried over Na2SO4 and concentrated under reduced pressure.
The resulting residue was purified by silica gel column chromatography (CHCl3/CH3OH=40/1 to 10/1) to obtain the title compound (120.3 mg, 95.1%) as a colorless solid. FAB-MS _m/z_ 509
(M+H)+ as C22H40N2O7S2; 1H NMR (400 MHz, CD3OD) δ 0.87–0.99 (m, 3H), 1.23–1.42 (m, 7H), 1.47 (s, 9H), 1.80–1.98 (m, 1H), 2.05–2.15 (m, 1H), 2.15 (s, 3H), 2.20–2.39 (m, 1H), 2.97 (br t,
_J_=9.6 Hz, 1H), 3.39–3.58 (m, 1H), 3.54 (dd, _J_=10.2, 3.0 Hz, 1H), 3.66 (dd, _J_=10.2, 7.4 Hz, 1H), 3.80–3.90 (m, 1H), 4.02–4.18 (m, 2H), 4.26–4.46 (m, 2H) and 5.25 (d, _J_=5.5 Hz, 1H).
7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-7-{(4-(PYRIDIN-3-YL)PHENYLTHIO}LINCOMYCIN (72) To a solution of 3-(4-bromophenyl)pyridine (75.4 mg, 0.32 mmol),
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (13.4 mg, 0.023 mmol) and tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3) (12.3 mg, 13.4 μmol) in 1,4-dioxane (1 ml) were added to
compound 71 (120.3 mg, 0.24 mmol) and _N_,_N_-diisopropylethylamine (82 μl, 0.47 mmol) and refluxed for 5 h. The mixture was filtrated by either Chromatodisc (0.45 μm) (Kurabo Industries) or
celite, concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH/28% aq. NH4OH=10/1/0.1) to obtain the title compound as an
off-white solid (125.9 mg, 80.4%). FAB-MS _m/z_ 662 (M+H)+ as C33H47N3O7S2; 1H NMR (400 MHz, CD3OD) δ 0.87–1.00 (m, 3H), 1.21–1.43 (m, 7H), 1.48 (s, 9H), 1.77–1.95 (m, 1H), 1.94, 1.97 (s x
2, 3H), 2.05–2.21 (m, 1H), 2.22–2.44 (m, 1H), 2.97 (t, _J_=9.6 Hz, 1H), 3.59 (dd, _J_=10.0, 3.2 Hz, 1H), 3.68 (br dd, _J_=9.8, 8.1 Hz, 1H), 3.85–4.01 (m, 2H), 4.05–4.16 (m, 1H), 4.28–4.41
(m, 1H), 4.41–4.50 (m, 1H), 4.51–4.64 (m, 1H), 5.27 (d, _J_=5.5 Hz, 1H), 7.47–7.57 (m, 3H), 7.59–7.69 (m, 2H), 8.05–8.14 (m, 1H), 8.51 (dd, _J_=4.8, 1.5 Hz, 1H) and 8.78–8.83 (m, 1H).
7(_S_)-1′-DEMETHYL-7-DEOXY-7-{(4-(PYRIDIN-3-YL)PHENYLTHIO}LINCOMYCIN (73) Compound 72 (125.9 mg, 0.19 mmol) and 2,2,2-trifluoroacetic acid (0.29 ml) in CH2Cl2 (2.5 ml) were treated at −20 °C
for 10 min, and then treated at 0 °C for 3 h according to the similar procedure as described for the preparation of 67 to afford 73 (99.1 mg, 92.7%) as a colorless solid. [α]D25 +91.4° (_c_
0.74, MeOH); ESI-MS _m/z_ 562 (M+H)+ as C28H39N3O5S2; TOF-ESI-HRMS (M+H)+ calcd. for C28H39N3O5S2: 562.2409, found: 562.2407; 1H NMR (400 MHz, CD3OD) δ 0.86–0.97 (m, 3H), 1.30–1.42 (m, 4H),
1.33 (d, _J_=6.9 Hz, 3H), 1.77–1.87 (m, 1H), 1.97 (s, 3H), 2.02–2.13 (m, 2H), 2.59 (dd, _J_=10.3, 8.0 Hz, 1H), 3.22 (dd, _J_=10.3, 7.0 Hz, 1H), 3.58 (dd, _J_=10.2, 3.3 Hz, 1H), 3.79 (br dd,
_J_=3.3, 0.9 Hz, 1H), 3.86 (dd, _J_=9.3, 3.7 Hz, 1H), 3.93 (dq, _J_=6.9, 2.6 Hz, 1H), 4.10 (dd, _J_=10.2, 5.6 Hz, 1H), 4.36–4.42 (m, 1H), 4.51 (dd, _J_=9.9, 2.6 Hz, 1H), 5.27 (d, _J_=5.6
Hz, 1H), 7.51 (ddd, _J_=8.0, 4.9, 0.9 Hz, 1H), 7.51–7.58 (m, 2H), 7.60–7.68 (m, 2H), 8.07 (ddd, _J_=8.0, 2.4, 1.6 Hz, 1H), 8.51 (dd, _J_=4.9, 1.6 Hz, 1H) and 8.79 (dd, _J_=2.4, 0.9 Hz, 1H).
7(_S_)-1′-_N_-(_TERT_-BUTOXYCARBONYL)-1′-DEMETHYL-7-DEOXY-7-{4-(PYRIMIDIN-5-YL)PHENYLTHIO}LINCOMYCIN (74) Compound 71 (116.4 mg, 0.23 mmol), 5-(4-bromophenyl)pyrimidine (107.3 mg, 0.46
mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (14.2 mg, 0.024 mmol), Pd2(dba)3 (11.8 mg, 0.013 mmol) and _N,N_-diisopropylethylamine (79.5 μl, 0.46 mmol) in 1,4-dioxane (1.5 ml)
were treated for 6 h according to the similar procedure as described for the preparation of 72 to afford 74 (117.9 mg, 77.7%) as a colorless solid. FAB-MS _m/z_ 663 (M+H)+ as C32H46N4O7S2;
1H NMR (400 MHz, CD3OD) δ 0.86–1.00 (m, 3H), 1.25–1.43 (m, 7H), 1.48, 1.47 (s x 2, 9H), 1.80–1.95 (m, 1H), 1.92, 1.95 (s x 2, 3H), 2.04–2.20 (m, 1H), 2.21–2.44 (m, 1H), 2.97 (t, _J_=9.6 Hz,
1H), 3.56–3.75 (m, 2H), 3.88–4.03 (m, 2H), 4.06–4.19 (m, 1H), 4.29–4.42 (m, 1H), 4.46 (d, _J_=9.6 Hz, 1H), 4.54–4.67 (m, 1H), 5.28 (d, _J_=5.5 Hz, 1H), 7.50–7.59 (m, 2H), 7.65–7.73 (m, 2H),
9.07 (s, 2H), 9.13 (s, 1H). 7(_S_)-1′-DEMETHYL-7-DEOXY-7-{4-(PYRIMIDIN-5-YL)PHENYLTHIO}LINCOMYCIN (75) Compound 74 (117.9 mg, 0.18 mmol) and 2,2,2-trifluoroacetic acid (0.27 ml) in CH2Cl2
(2.5 ml) were treated at −20 °C for 20 min, and then treated room temperature for 5 h according to the similar procedure as described for the preparation of 73 to afford 75 (82.2 mg, 82.1%)
as a colorless solid. [α]D25 +91.7° (_c_ 0.33, MeOH); ESI-MS _m/z_ 563 (M+H)+ as C27H38N4O5S2; TOF-ESI-HRMS (M+H)+ calcd. for C27H38N4O5S2: 563.2362, found: 563.2356; 1H NMR (400 MHz, CD3OD)
δ 0.78–0.88 (m, 3H), 1.20–1.32 (m, 4H), 1.25 (d, _J_=6.9 Hz, 3H), 1.68–1.78 (m, 1H), 1.86 (s, 3H), 1.92–2.05 (m, 2H), 2.55 (dd, _J_=10.2, 8.0 Hz, 1H), 3.12 (dd, _J_=10.2, 6.7 Hz, 1H), 3.48
(dd, _J_=10.2, 3.3 Hz, 1H), 3.69 (br dd, _J_=3.3, 0.7 Hz, 1H), 3.75 (dd, _J_=9.1, 3.4 Hz, 1H), 3.87 (dq, _J_=6.9, 2.6 Hz, 1H), 4.01 (dd, _J_=10.2, 5.5 Hz, 1H), 4.27–4.32 (m, 1H), 4.43 (dd,
_J_=9.9, 2.6 Hz, 1H), 5.18 (d, _J_=5.5 Hz, 1H), 7.43–7.50 (m, 2H), 7.55–7.65 (m, 2H), 8.97 (s, 2H), 9.03 (s, 1H). _IN VITRO_ ANTIBACTERIAL ACTIVITY MIC (μg ml−1) was determined by the agar
dilution method, which was described in Clinical and Laboratory Standards Institute (M07-06 in 2003). Test strains of _S. pneumoniae_ and _S. pyogenes_ were subjected to seed culture using
brain heart infusion agar (Becton Dickinson and Company, Tokyo, Japan) and 5% defibrinated horse blood. Test strains of _H. influenzae_ were subjected to seed culture using sensitivity disk
agar-N ‘Nissui’ (Nissui, Tokyo, Japan), 5% defibrinated horse blood, 5 μg ml−1 Hemin and 15 μg ml−1 nicotinamide adenine dinucleotide. A 5 μl portion of cell suspension of the test strains
having about 106 colony-forming unit per ml was inoculated into sensitivity disk agar-N ‘Nissui’ supplemented with 5% defibrinated horse blood, 5 μg ml−1 Hemin and 15 μg ml−1 nicotinamide
adenine dinucleotide, and incubated at 37 °C for 18–22 h. Then, the MIC was measured. REFERENCES * Reinert, R. R., van der Linden, M. & Al-Lahham, A. Molecular characterization of the
first telithromycin-resistant _Streptococcus pneumoniae_ isolate in Germany. _Antimicrob. Agents Chemother._ 49, 3520–3522 (2005). Article CAS Google Scholar * Kim, S. H. _et al_.
Changing trends in antimicrobial resistance and serotypes of _Streptococcus pneumoniae_ isolates in Asian countries: an Asian Network for Surveillance of Resistant Pathogens (ANSORP) study.
_Antimicrob. Agents Chemother._ 56, 1418–1426 (2012). Article CAS Google Scholar * Ajito, K., Miura, T., Furuuchi, T. & Tamura, A. Sixteen-membered macrolides: chemical modifications
and future applications. _Heterocycles_ 89, 281–352 (2014). Article CAS Google Scholar * Morimoto, S., Takahashi, Y., Watanabe, Y. & Omura, S. Chemical modification of erythromycins.
I. Synthesis and antibacterial activity of 6-_O_-methylerythromycins A. _J. Antibiot._ 37, 187–189 (1984). Article CAS Google Scholar * Slobodan, D. _et al_. Erythromycin series. Part 13.
Synthesis and structure elucidation of 10-dihydro-10-deoxo-11-methyl-11-azaerythromycin A. _J. Chem. Res. Synop._ 152–153 (1988). * Retsema, J. _et al_. Spectrum and mode of action of
azithromycin (CP-62,993), a new 15-membered-ring macrolide with improved potency against Gram-negative organisms. _Antimicrob. Agents Chemother._ 31, 1939–1947 (1987). Article CAS Google
Scholar * Denis, A. _et al_. Synthesis and antibacterial activity of HMR 3647 a new ketolide highly potent against erythromycin-resistant and susceptible pathogens. _Bioorg. Med. Chem.
Lett._ 9, 3075–3080 (1999). Article CAS Google Scholar * Clay, K. D. _et al_. Severe hepatotoxicity of telithromycin: three case reports and literature review. _Ann. Intern. Med._ 144,
415–420 (2006). Article Google Scholar * Ross, D. B. The FDA and the case of ketek. _N. Engl. J. Med._ 356, 1601–1604 (2007). Article CAS Google Scholar * Gleason, P. P., Walters, C.,
Heaton, A. H. & Schafer, J. A. Telithromycin: the perils of hasty adoption and persistence of off-label prescribing. _J. Manag. Care Pharm._ 13, 20–25 (2007). Google Scholar *
Department of Health and Human Services. Telithromycin (marketed as ketek) information. Available at:
http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm107824.htm (last accessed 26 April 2007). * Miura, T. _et al_. Novel azalides derived from
sixteen-membered macrolides. I. Isolation of the mobile dialdehyde and its one-pot macrocyclization with an amine. _J. Antibiot._ 60, 407–435 (2007). Article CAS Google Scholar * Miura,
T. _et al_. Novel azalides derived from 16-membered macrolides. III. Azalides modified at the C-15 and 4″ positions: improved antibacterial activities. _Bioorg. Med. Chem._ 18, 2735–2747
(2010). Article CAS Google Scholar * Mason, D. J., Dietz, A. & Deboer, C. Lincomycin, a new antibiotic I. Discovery and biological properties. _Antimicrob. Agents Chemother._ 1962,
554–559 (1962). Google Scholar * Magerlein, B. J. & Lincomycin, X. The chemical synthesis of lincomycin. _Tetrahedron Lett._ 1, 33–36 (1970). Article Google Scholar * Howarth, G. B.,
Szarek, W. A. & Jones, J. K. N. The synthesis of lincomycin. _J. Chem. Soc. (c)_ 16, 2218–2224 (1970). CAS Google Scholar * Perlman, D. _Structure–Activity Relationships Among the
Semisynthetic Antibiotics_ 600–651 (Harcourt Brace Jovanovich Publishers and Academic Press, New York, San Francisco, London, 1977). * Birkenmeyer, R. D. & Kagan, F. Lincomycin. XI.
Synthesis and structure of clindamycin. A potent antibacterial agent. _J. Med. Chem._ 13, 616–619 (1970). Article CAS Google Scholar * Shan, P. J., Vakil, N. & Kabakov, A. Role of
intravenous immune globulin in streptococcal toxic shock syndrome and _Clostridium difficile_ infection. _Am. J. Health Syst. Pharm._ 72, 1013–1019 (2015). Article Google Scholar *
Hoeksema, H. in Abstr. Pap. Division of Carbohydrate Chemistry, 149th Meet. Am. Chem. Soc. Detroit, MI, 9C (The Upjohn Company, Kalamazoo, MI, 1965. * Magerlein, B. J., Birkenmeyer, R. D.
& Kagan, F. Chemical modification of lincomycin. _Antimicrob. Agents Chemother._ 6, 727–736 (1966). CAS PubMed Google Scholar * Sinkula, A. A., Morozowich, W., Lewis, C. &
Mackellar, F. A. Synthesis and bioactivity of lincomycin-7-monoesters. _J. Pharm. Sci._ 58, 1389–1392 (1969). Article CAS Google Scholar * Magerlein, B. J. & Kagan, F. Lincomycin. IX.
7-Thiol and thioamido analogs of lincomycin. _J. Med. Chem._ 12, 974–977 (1969). Article CAS Google Scholar * Lewis, J. G. _et al_ in 43rd Interscience Conference on Antimicrobial Agents
and Chemotherapy, Poster F-1388 (Washington, DC, USA, 2004). * Bannister, B. Modifications of lincomycin involving the carbohydrate portion. Part III. The 7-_O_-methyl and
6-de-(1-hydroxyethyl) analogues. _J. Chem. Soc. Perkin Trans. I_ 16, 1676–1682 (1973). Article CAS Google Scholar * Bannister, B. Modifications of lincomycin involving the carbohydrate
portion. Part IV. (7_S_-7-alkoxy-7-deoxy-analogues. _J. Chem. Soc. Perkin Trans. I_ 3, 360–369 (1974). Article CAS Google Scholar * Bannister, B. & Mydlow, P. K. The S-alkylation of
sulphides by an activated carbohydrate epimine under acidic catalysis: The formation of α-acetamido-sulphides. Part 5. The introduction of functionality into the sulphide substituent. _J.
Chem. Res. (S)_ 1989, 90–91 (1989). Google Scholar * Bannister, B. The S-alkylation of sulphides by an activated carbohydrate epimine under acidic catalysis: the formation of
α-acetamido-sulphides. Part 4. Reaction with dithioacetals and monothioacetals. _J. Chem. Soc. Perkin. Trans. I_ 1980, 540–552 (1980). Article Google Scholar * Bannister, B.
(7_S_-7-deoxy-7-substituted-alkylthio-lincomycin. _S_-Alkylation of sulphides by an activated epimine under acidic catalysis: formation of α-acetamido-sulphides. _Tetrahedron_ 40, 1633–1660
(1984). Article CAS Google Scholar * Bannister, B. Derivatives of lincomycin and its analogs and process. US Patent US3915954 A (The Upjohn Company, 1973). * Bannister, B. Derivatives of
lincomycin and its analogs and process. Canadian Patent CA-971956 A1 (The Upjohn Company, 1972). * Sztaricskai, F. _et al_. Semisynthetic modification of antibiotic lincomycin. _J.
Antibiot._ 49, 941–943 (1996). Article CAS Google Scholar * Umemura, E. _et al_ Lincomycin derivative and antibacterial agent containing the same as active ingredient. Japanese patent
WO/2007/066805 A1 (14 June 2007). * Wakiyama, Y. _et al_ Lincomycin derivatives and antibacterial agents containing the same as the active ingredient. Japanese patent: WO/2008/146917 A1 (4
December 2008). * Umemura, E. _et al_ Lincosamide derivative, and antibacterial agent comprising the same as active. Japanese patent WO/2008/146919 A1 (4 December 2008). * Umemura, E. _et
al_. Synthesis of novel lincomycin derivatives and their _in vitro_ antibacterial activities. _J. Antibiot._ 66, 195–198 (2013). Article CAS Google Scholar * Wakiyama, Y. _et al_.
Synthesis and structure–activity relationships of novel lincomycin derivatives. Part 1. Newly generated antibacterial activities against Gram-positive bacteria with erm gene by C-7
modification. _J. Antibiot._ 69, 368–380 (2016). Article CAS Google Scholar * Wakiyama, Y. _et al_. Synthesis and structure–activity relationships of novel lincomycin derivatives. Part 2.
Synthesis of 7(_S_-7-deoxy-7-(4-morpholinocarbonylphenylthio)lincomycin and its 3-dimensional analysis with rRNA. _J. Antibiot._ 69, 428–439 (2016). Article CAS Google Scholar * Kumura,
K. _et al_. Synthesis and antibacterial activity of novel lincomycin derivatives. I. Enhancement of antibacterial activities by introduction of substituted azetidines. _J. Antibiot._ 69,
440–445 (2016). Article CAS Google Scholar * Wakiyama, Y. _et al_. Synthesis and structure–activity relationships of novel lincomycin derivatives part 3: discovery of the
4-(pyrimidin-5-yl)phenyl group in synthesis of 7(_S_-thiolincomycin analogs. _J. Antibiot._ 70, 52–64 (2017). Article CAS Google Scholar * Kumura, K. _et al_. Synthesis and antibacterial
activity of novel lincomycin derivatives. II. Exploring (7_S_-7-(5-aryl-1,3,4-thiadiazol-2-yl-thio)-7-deoxylincomycin derivatives. _J. Antibiot_ (e-pub ahead of print 7 December 2016;
doi:10.1038/ja.2016.139)). * Argoudelis, A. D., Coats, J. H., Mason, D. J. & Sebek, O. K. Microbial transformation of antibiotics. III. Conversion of clindamycin to
1′-demethylclindamycin and clindamycin sulfoxide by Streptomyces species. _J. Antibiot._ 22, 309–314 (1969). Article CAS Google Scholar * Magerlein, B. J., Birkenmeyer, R. D. & Kagan,
F. Lincomycin. VI. 4′-alkyl analogs of lincomycin. Relationship between structure and antibacterial activity. _J. Med. Chem._ 10, 355–359 (1967). Article CAS Google Scholar * Magerlein,
B. J. Lincomycin. 14. An improved synthesis and resolution of the antimalarial agent, 1′-demethyl-4′-depropyl-4′ (_R_- and -(_S_-pentylclindamycin hydrochloride (U-24,729A). _J. Med. Chem._
15, 1255–1259 (1972). Article CAS Google Scholar * Magerlein, B. J. Lincomycin. VII. 4′-depropyl-4′-ethoxylincomycins. _J. Med. Chem._ 10, 1161–1163 (1967). Article CAS Google Scholar
* Lewis, J. G. _et al_ Novel lincomycin derivatives possessing antimicrobial activity. Japanese patent WO/2006/055070 A2 (26 May 2006). * Schlünzen, F. _et al_. Structural basis for the
interaction of antibiotics with the peptidyl transferase centre in eubacteria. _Nature_ 413, 814–821 (2001). Article Google Scholar * Pedregal, C., Ezquerra, J., Escribano, A., Carreńo, M.
C. & Ruano, J. G. Highly chemoselective reduction of _N_-Boc protected lactams. _Tetrahedron Lett._ 35, 2053–2056 (1994). Article CAS Google Scholar * Llinàs-Brunet, M. _et al_.
Highly potent and selective peptide-based inhibitors of the hepatitis C virus serine protease: towards smaller inhibitors. _Bioorg. Med. Chem. Lett._ 10, 2267–2270 (2000). Article Google
Scholar * Itoh, T. & Mase, T. A general palladium-catalyzed coupling of aryl bromides/triflates and thiols. _Org. Lett._ 6, 4587–4590 (2004). Article CAS Google Scholar Download
references ACKNOWLEDGEMENTS We thank A Tamura, Dr E Shitara and Dr T Yoshida for encouragement and valuable discussion. We are grateful to Professor Emeritus Dr M Konno for supervision
through our in-house drug discovery program in lincomycin field. We also thank M Ishii for direction in intellectual properties, T Miyara, S Miki and K Kaneda for analytical and synthetic
chemistry, Y Takayama and K Yamada for biological studies, and M Takagi for English manuscript. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Pharmaceutical Research Center, Meiji Seika
Pharma, Yokohama, Japan Yoshinari Wakiyama, Ko Kumura, Eijiro Umemura, Kazutaka Ueda, Takashi Watanabe, Keiko Yamada, Takafumi Okutomi & Keiichi Ajito Authors * Yoshinari Wakiyama View
author publications You can also search for this author inPubMed Google Scholar * Ko Kumura View author publications You can also search for this author inPubMed Google Scholar * Eijiro
Umemura View author publications You can also search for this author inPubMed Google Scholar * Kazutaka Ueda View author publications You can also search for this author inPubMed Google
Scholar * Takashi Watanabe View author publications You can also search for this author inPubMed Google Scholar * Keiko Yamada View author publications You can also search for this author
inPubMed Google Scholar * Takafumi Okutomi View author publications You can also search for this author inPubMed Google Scholar * Keiichi Ajito View author publications You can also search
for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Keiichi Ajito. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. RIGHTS AND
PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Wakiyama, Y., Kumura, K., Umemura, E. _et al._ Synthesis and structure–activity relationships of novel lincomycin
derivatives. Part 4: synthesis of novel lincomycin analogs modified at the 6- and 7-positions and their potent antibacterial activities. _J Antibiot_ 70, 888–906 (2017).
https://doi.org/10.1038/ja.2017.54 Download citation * Received: 18 January 2017 * Accepted: 20 March 2017 * Published: 31 May 2017 * Issue Date: August 2017 * DOI:
https://doi.org/10.1038/ja.2017.54 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently
available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative