Improved group-specific PCR primers for denaturing gradient gel electrophoresis analysis of the genetic diversity of complex microbial communities
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Phylum- and class-specific PCR primers were tested for the production of clone libraries and for denaturing gradient gel electrophoresis (DGGE) analysis of complex bacterial communities.
Primers were designed to specifically amplify 16S rRNA gene fragments of the phyla Bacteroidetes, Planctomycetes and Firmicutes, of three classes of the phylum Proteobacteria, the
Alphaproteobacteria, Betaproteobacteria and Gammaproteobacteria, and of the Cyanobacteria (including chloroplast 16S rRNA genes). The specificity of the seven primer pairs was tested by
producing clone libraries from environmental DNA samples from mesotrophic (Norwegian coastal) and oligotrophic (Northern Atlantic Gyre) environments. Five of the seven primer pairs
specifically amplified target 16S rRNA gene sequences. Exceptions were the Betaproteobacteria- and Firmicutes-specific primers, which were relatively successful with coastal water mesocosm
samples but less so with the Northern Atlantic Gyre sample. Phylogenetic analysis of sequences from the Gammaproteobacteria clone library revealed that the coastal sample yielded a number of
clones that clustered within clades that belong to the oligotrophic marine Gammaproteobacteria (OMG) group, indicating that this group is not confined exclusively to the oligotrophic
environment. Comparison of the bacterial diversity of the environmental DNA sample from the coastal and the open ocean using a two- or three-step nested PCR-DGGE process revealed significant
differences in the bacterial communities. The application of the group-specific primers provides a higher resolution genetic fingerprinting approach than existing DGGE primer sets.
The study of biodiversity of marine microbial communities has become commonplace in microbial ecology, with the 16S ribosomal RNA (rRNA) gene being the most frequently used phylogenetic
marker (Amann et al., 1995; Pernthaler and Amann, 2005). This marker gene has revealed the great genetic diversity of bacteria that is now assembled in the Ribosomal Database Project-II
(RDP-II; http://rdp.cme.msu.edu/). This currently stores approximately 418 000 partial 16S rRNA gene sequences, though it is likely that there is some degree of redundancy and sequence
anomalies within the database (Ashelford et al., 2005). This genetic diversity also means that it is very unlikely that the whole range of diversity can be detected in a single sample,
especially where communities are complex. Attempts to estimate the total bacterial community diversity have used partial analysis of the total community (that is clone library screening)
combined with theoretical models. This approach, however, does not reveal the identity of the less abundant components of the assemblage. Increasing the number of clones per clone library
has been successful in detecting novel bacterial clades (for example, Chouari et al., 2005) or comparing different marine environments in terms of bacterial community composition (for
example, Rappé et al., 2000; Zaballos et al., 2006). However, despite the decreasing costs for nucleotide sequencing, the success of this approach is still limited because of the huge scale
of bacterial diversity — perhaps as many as 2 × 106 different species in the oceans (Curtis et al., 2002). The recent arrival of pyrosequencing of 16S rRNA tags (Sogin et al., 2006) may
represent an alternative because of the much lower cost per sequence; but pyrosequencing still does not allow analyses and comparison of the bacterial diversity in different environments on
a routine basis.
Alternative approaches, such as denaturing gradient gel electrophoresis (DGGE), are routinely used to determine diversity because they avoid large-scale sequencing efforts. However, these
are also likely to detect only a small subfraction of the total diversity. The use of bacterial PCR primers is likely to miss minor fractions of the microbial community because most of the
PCR product will be composed of the more abundant species. Faint DNA bands on DGGE gels are unlikely to be detected or their identity determined. To overcome this limitation and to detect
less abundant sequence clones, Holben et al. (2004) fractionated 16S rRNA gene sequences from a microbial community according to their G+C content before DGGE analysis. However, with the
possible exception of the high G+C-containing Actinobacteria, this method has limited application and does not separate bacteria by phylogeny. Combining bromodeoxyuridine immunocapture and
DGGE has been proposed to separate the DNA of the actively growing bacteria from the rest of the environmental DNA (Hamasaki et al., 2007). However, this results in the analysis of subgroups
of bacteria that are not defined on phylogenetic criteria, and does not allow screening of the whole range of genetic bacterial diversity.
An alternative approach is the application of group-specific oligonucleotide probes. This has mainly been used in combination with fluorescence in situ hybridization (FISH). FISH has been a
very effective technique but, in contrast to the analysis of clone libraries produced from PCR fragments, FISH cannot reveal unknown bacterial groups. It only detects and quantifies those
bacteria that the probes were designed to detect. Group-specific PCR primers have already been used for the analysis of complex microbial communities. For example, Mehling et al. (1995)
designed and used PCR primers for the 16S rRNA gene sequences of Streptomycetes. Driven by the biotechnological potential of compounds and activities found in Actinobacteria in general,
Stach et al. (2003) developed new primers to detect a wide range of Actinobacteria in terrestrial and marine environments. Cyanobacterial and chloroplast 16S rRNA gene fragments can also be
amplified specifically (Nübel et al., 1997). PCR primers have also been designed to amplify more defined groups of bacteria, such as those of the genus Pseudomonas (for example, Widmer et
al., 1998). Many studies on pathogens have focused on well-defined species or strains of pathogens in the environment (for example, Miyamoto et al., 1997). Other microbial ecology studies
have involved PCR primers specific for functional groups, such as the ammonia-oxidizing bacteria (McCaig et al., 1994; Freitag and Prosser, 2003; Calvo et al., 2004) and sulphate-reducing
bacteria (Stubner, 2004; Dar et al., 2005). The study by Dar et al. (2005) provides also a convincing case that group-specific primers can be used in combination with DGGE to detect less
abundant bacterial groups in a complex community. The novelty of that approach was to use group-specific primers in a PCR followed by reamplification of the PCR product with nested,
universal bacterial DGGE primer. This overcomes the difficulty in designing novel DGGE primers and establishing the optimal conditions for those primers (Dar et al., 2005).
As 16S rRNA gene sequence databases continue to grow, group-specific PCR primers must be continually re-evaluated for their specificity and range of sequence matches (Baker et al., 2003).
Furthermore, new and more sophisticated software packages, such as PRIMROSE (Ashelford et al., 2002) and ARB (Ludwig et al., 2004), can help researchers to design 16S rRNA gene probes. These
software packages use different sequence databases and each has its specific strengths. For example, as well as differences in the algorithm used for searching for priming sites, it is
possible to use PRIMROSE to design degenerate probes, a function that is not available in ARB. ARB, however, allows the search for probes to be based on a greater number of specific
parameters.
To analyse the total bacterial community during changing environmental conditions and seasons, we have developed group-specific primers for seven different taxonomic groups. Apart from
expensive large-scale sequencing of clone libraries, DGGE remains one of the few fingerprinting techniques currently available that allows analysis of the whole microbial community and
identification, by sequence analysis of DGGE bands, of specific members of that community. Therefore, we used a similar DGGE approach to that of Dar et al. (2005), that is reamplification of
the bacterial group-specific PCR products with a nested or semi-nested universal DGGE-PCR primer set. We tested these primers for their specificity and applicability by sequence analysis of
clone libraries and comparative DGGE analysis of total environmental DNA from a coastal nutrient-rich and an oligotrophic open ocean environment.
PCR primers were designed using the ARB (Ludwig et al., 2004) and PRIMROSE (Ashelford et al., 2002) programs; the latter is part of the TOOLKIT software package
(http://www.cf.ac.uk/biosi/research/biosoft). The name given to each primer consists of a short name indicating the target group, followed by a number, representing the position of the first
base of the primer within the Escherichia coli 16S rRNA gene sequence and ‘f’ or ‘r’ indicating whether the primer is the forward or reverse primer, respectively. The sequences of the
primers and their specificity are summarized in Table 1. The number of target and non-target matches of the primers was tested in silico using the PROBE MATCH function within the RDP-II
database (http://rdp.cme.msu.edu) and the ROSE function of TOOLKIT which identifies the number of sequences, among the 15 104 Bacterial sequences in its database, to which the primer
sequence is homologous.
A nested Bacteria primer pair was selected for each group-specific primer pair to be used in a nested re-PCR (Table 2). Only one nested Bacteria primer was available in the cases of the
Betaproteobacteria-, the Bacteroidetes- and the Cyanobacteria/chloroplast-specific primers. Therefore, one of the group-specific primers was paired with the Bacteria primer for a semi-nested
re-PCR (Table 2), and a GC clamp was attached to the 5′-position of one of the two primers (indicated in the primer name with ‘-GC’; Table 2).
Samples from two contrasting environments were used in this study — a mesotrophic coastal and an oligotrophic open ocean environment. The coastal sample was collected in May 2003 from a
20-m3 mesocosm after 12 days of incubation during the ‘Pelagic Ecosystem CO2 Enrichment Study’ (PeECE; http://peece.ifm-geomar.de/) at the EU Large-Scale-Facility, University of Bergen,
Norway, located at Espeland in the Raunefjord (60.27°N 5.22°E), 20 km south of Bergen. During the experiment, nitrate, phosphate and silicate were added at typical winter concentrations to
stimulate a phytoplankton bloom. The open ocean, oligotrophic sample was obtained from 15 m depth in the Northern Atlantic Gyre (35°N 20°W) collected during the Atlantic Meridional Transect
(AMT-15) cruise in September 2004 (Robinson et al., 2006).
Samples (4 l) from the mesocosm and samples (7 l) from the Northern Atlantic Gyre, respectively, were filtered through Sterivex cartridges with 0.2 μm pore size filter membrane (Millipore,
Watford, UK). The cartridges were stored at −70 °C until analysis. Total environmental DNA was isolated from the cartridges using the method described by Jameson et al. (2007). This involved
lysozyme treatment with 1.5 ml of lysis buffer (200 mM Tris-HCl, pH 8.0; 1.5 mg lysozyme; 125 mM EDTA), incubated at 37 °C on a rotary shaker (10 r.p.m.) for 30 min, with a 90° turn of the
cartridge every 6 min to ensure coverage of the whole inner surface area of the cartridge. After addition of 0.25 ml of proteinase K lysis buffer (to a final concentration of 1.25% w/v SDS,
300 μg proteinase K, 70 mM NaCl), the cartridge was incubated at 37 °C for 15 min followed by another 60 min at 55 °C, again with regular rotation of the cartridge. Subsequently, the lysis
mix was transferred to microcentrifuge tubes and extracted twice with one volume of chloroform/isopropanol (24:1). The DNA was precipitated (1 vol isopropanol; 0.4 vol 7.5 M ammonium
acetate), and the pellet was washed with 70% (v/v) ethanol. The DNA pellet was resuspended in sterile, double-distilled water.
Clone libraries were prepared from PCR-amplified 16S rRNA gene fragments for each of the group-specific primer pairs. A nested PCR approach was required for three of the primer pairs to
obtain sufficient PCR product for subsequent cloning (see Table 1). In these cases, aliquots of the PCR products obtained with the Bacteria primer pair 9bfm/1512uR were used as templates for
a reamplification with a nested group-specific primer set. A typical PCR reaction was carried out in 25-μl volumes and contained 2 mmol l−1 MgCl2, 200 μmol l−1 dNTPs, 2.5 U of Taq DNA
polymerase (Promega, Southampton, UK) and 50 nmol l−1 of each of the primers. The temperatures for primer annealing were dependent on the primer pair used (summarized in Table 1). They were
determined using the equation Tm=(4 × GC)+(2 × AT). A range of annealing temperatures (ATs) similar to the calculated Tm were then tested in a gradient PCR to determine empirically the
temperature that resulted in the most specific PCR product. All PCRs used the same basic cycle protocol except for the AT: following an initial denaturation step of 4 min at 96 °C, 30 PCR
cycles were performed (96 °C for 60 s, AT for 60 s, 74 °C for 60 s) followed by a final extension step at 74 °C for 10 min. The extension time was increased to 90 s for primer pair
9bfm/1512uR. In all cases, the PCR yielded only specific products, that is single bands as judged by electrophoresis of the PCR products on agarose gels.
Aliquots of the products from PCRs or re-PCRs were cloned into a TA vector using the pGEM-T Easy Vector System I cloning kit (Promega). Twenty or 50 clones were picked from each of the
group-specific clone libraries prepared from the mesocosm and the Northern Atlantic Gyre samples, respectively, and the 16S rRNA gene fragments were reamplified using vector primers (M13).
The PCR products were used for the sequencing reactions.
PCR products were treated with ExoSapIT (Amersham Biosciences, Little Chalfont, UK) according to the manufacturer's instructions and used directly for sequence analysis. Nucleotide
sequencing was performed using the BigDye Terminator v3.1 cycle sequencing kit (ABI). An M13 primer was used in the cycle sequencing reaction. Sequences were analysed on an ABI 3100
automatic sequencer. Generally, only one strand of the DNA fragments was sequenced. This proved to be sufficient for the taxonomic identification of the cloned 16S rRNA gene fragments using
the BLAST search function within the NCBI database. Both DNA strands were sequenced in those cases where sequences could not be assigned easily to taxonomic groups or where the quality of
the nucleotide sequence could not be determined unambiguously.
Denaturing gradient gel electrophoresis (DGGE) was based on Muyzer et al. (1998) and Dar et al. (2005). In essence, group-specific PCR products were used as template in a re-PCR with a
nested Bacteria DGGE-PCR primer pair (Table 2). Semi-nested re-PCRs were used in the case of the Cyanobacteria/chloroplasts, the Bacteroidetes and the Betaproteobacteria (Table 2). Only one
bacterial primer was available within the 16S rRNA gene fragment that was amplified by the primers specific for these phylogenetic groups. PCR conditions and cycle protocol for the nested
PCRs with the DGGE primer pairs were the same as those used for the PCRs with the group-specific primer pairs, except for the ATs which are shown in Table 2.
DGGE of the PCR products was performed on a 8% (w/v) polyacrylamide gel with urea and formamide as denaturants. The denaturing gradients varied with the DGGE primer pair used for the PCR but
were generally between 40% and 60% (Table 2). Electrophoresis was performed in 1 × Tris-acetate EDTA buffer at 60 °C at constant voltage of 60 V for 18 h. Subsequently, gels were stained in
1 × SYBR Gold nucleic acid gel stain (Molecular Probes) for 45 min and rinsed in distilled water prior to image analysis on a Syngene GelDoc station. Individual DGGE bands were cut out from
the gel and incubated overnight at 4 °C in 30 μl H2O. An aliquot (ca 5 μl) was used in a PCR with the same primer set used for DGGE (Table 2; but without the GC clamp attached to one
primer) to reamplify the insert. The nucleotide sequences of these 16S rRNA gene fragments were determined as described above.
Sequences from the clone library prepared with the Gammaproteobacteria-specific primers were first compared to sequences stored in GenBank using the BLAST algorithm. Subsequently, the
sequences from the clone libraries, and sequences with high similarity as identified by the BLAST searches, were imported into the ARB software program
(http://www.mikro.biologie.tu-muenchen.de/pub/ARB) and aligned to other Gammaproteobacteria 16S rRNA gene sequences using the automated alignment tool within ARB (Ludwig et al., 2004). The
alignment was further corrected ‘by eye’, taking the secondary structure prediction of the ARB program into account. Phylogenetic analyses were based on the alignment of the 16S rRNA gene
sequences from the mesocosm and Northern Atlantic Gyre clone libraries with representative sequences from the sequence databases. Calculation of the phylogenetic trees was based on these
sequence alignments using the neighbour-joining method with Jukes–Cantor corrections, as well as the maximum likelihood and parsimony algorithms. Analyses were carried out using a maximum
frequency filter. For each of the phylogenetic analyses in this study, the grouping of strains and environmental clones within the different clusters of the tree was identical for all of the
above three phylogenetic methods for calculating trees (maximum likelihood, maximum parsimony, neighbour-joining). However, those branching points within a tree that were not supported by
each of the three algorithms were collapsed within the neighbour-joining tree using a strict consensus rule until the branching was supported in all three analyses. The neighbour-joining
tree was chosen for depicting the phylogenic relationship of the 16S rRNA gene clones and strains, and bootstrap values were calculated from 100 trees using the neighbour-joining method.
The sequence data of 16S rRNA gene fragments have been submitted to the EMBL database with accession numbers AM706671-707020 (Northern Atlantic Gyre clone library), AM706537-AM706670
(mesocosm clone library), AM747394-AM747468 (sequences of DGGE bands).
Blackwood et al. (2005) used ARB to develop five group-specific primers including primers for four of the groups of bacteria investigated in this study. Ashelford et al. (2002) compared in
detail the PRIMROSE and ARB programs and concluded that in many cases it was possible to identify better oligonucleotide probes (judged by in silico analysis) using PRIMROSE rather than ARB.
We based the development of our primers on the independent use of both software packages to ensure the best design. The theoretical specificities of all primers were tested with the ROSE
program of the TOOLKIT software package, as well as the PROBE MATCH function within the RDP database (http://rdp.cme.msu.edu/). The sequences of the primers and their specificity are
summarized in Table 1. For the purpose of comparison (see below under ‘Group-specific clone libraries’), we preferred the use of the ROSE program of the TOOLKIT software package, since this
uses exactly the same sequences as a basis for the comparison. In contrast, results of sequence searches in the current version of the PROBE MATCH function within the RDP database depend on
the search window set for the priming site, which is different for different primer locations along the 16S rRNA gene.
PCR primers were developed for the amplification of 16S rRNA gene fragments that provide valuable additions to existing primers. In silico analyses indicate that they have generally a higher
number of exact matches to the 16S rRNA gene sequences from members of the target group of bacteria for which they were designed, while their specificity is generally similar to that of
published primers (Table 3).
However, some of the most suitable primers were identical to the FISH probes suggested by Ashelford et al. (2002)—Alf28f, Beta359f and Beta682r. Other probes suggested by Ashelford et al.
(2002) or previously published primers (for example, Nübel et al., 1997; Blackwood et al., 2005) exploit similar priming sites to those used in our study but were, for example, of different
length (for example, Alf684r, CFB555 and CYA785r) and degeneracy (for example, CYA361f). Blackwood et al. (2005) and Blümel et al. (2007) paired their group-specific primers with either the
universal primer 1392r or the Bacteria primers Eub338 or 27f. In contrast, we designed two group-specific primers (that is a primer pair) for each taxonomic group to increase specificity of
the PCR, or to ensure that a more group-specific primer of the primer pair would compensate for lower specificity of the other primer (for example, CFB555f/CFP968r compared to 27F/Cyt1020R
of Blümel et al., 2007).
The group-specific primer pairs were generally used to amplify 16S rRNA gene fragments directly from environmental DNA. However, primer pairs for the Alphaproteobacteria, the Planctomycetes
and the Firmicutes resulted in only low yield when environmental DNA was used as template. Therefore, a nested PCR approach was designed to overcome this problem. Obviously, the forward
primer used in the first PCR had to be upstream of the Alphaproteobacteria-specific forward primers (Alf28f), which were located close to the 5′-end of the 16S rRNA gene. Concerns have been
raised for some time when the bacterial forward primers 8f and 27F are used because of limited amplification efficiency and potential mismatches with newly discovered strains or
environmental 16S rRNA gene sequences (for example, Marchesi et al., 1998). These primers were designed when the sequence databases consisted only of a few thousand clones (for example, see
under http://rdp.cme.msu.edu/misc/history.jsp). Testing Bacteria primers for specificity revealed that the sequences were homologous to only 56.5% (8f; Hicks et al., 1992) or 72.9% (27F;
Giovannoni et al., 1996) of the over 15 000 sequences within the PRIMROSE database. The sequence of the universal (bacteria and archaea) reverse primer 1512uR (Weisburg et al., 1991) was
homologous to 78.5% of the sequences tested (Table 1).
We decided to use reverse primer 1512uR but modify the forward primer to account for sequence differences at this priming site. In silico analysis showed that the sequence of the new
Bacteria primer 9bfm (Table 1) is homologous to 77.7% of the bacterial sequences in the PRIMROSE database but also to one Archaeon (Methanobrevibacter sp strain MB-9; accession AB017514).
However, an archaeal 16S rRNA gene sequence was never detected, either in any of the libraries screened in this study (see below) or in any other analyses in our laboratory. Although there
is only a small increase (5%) in the number of sequences to which the sequence of primer 9bfm is homologous, this is likely to lead to a far higher amplification rate. The greater degeneracy
of the primer is likely to result in the successful amplification of more novel and yet unknown genotypes since many of the sequences that are not homologous to primer 9bfm will differ only
in one base (as tested using the OLIGOCHECK function within the TOOLKIT software package). Furthermore, this one-base difference is not within the last three or four 3′-end bases, which are
important for the polymerase to start extending the DNA strand during PCR.
However, despite the high percentage of exact matches to bacterial 16S rRNA gene sequences, primers may still be biased against certain groups of bacteria while matching the 16S rRNA gene
sequence of most strains of other phylogenetic groups. Therefore, primers should always be tested with respect to the specific bacterial group of interest prior to use.
In contrast to group-specific primers (see below), the application of these general Bacteria primers aims to amplify the whole genetic diversity of the domain. Therefore, use of less
stringent conditions in the PCR reactions, for example a lower AT than the melting temperature of the primer, will allow for mismatches during the annealing, thus overcoming some of the
above-mentioned biases. This further increases the genetic range of bacterial 16S rRNA gene fragments that are amplified in a PCR. In practice, it was found that, despite the increased
degeneracy of primer 9bfm and the lower than optimal AT used in our PCRs (52 °C instead of ⩾54 °C Tm), all PCRs resulted in specific amplification of 16S rRNA gene fragments as judged by
sequence analyses of clone libraries prepared with these primers (unpublished data). It should be noted that none of the PCRs with this primer pair in our laboratory has yet led to the
amplification of nonspecific products.
The newly developed primers were tested with environmental DNA samples from contrasting environments—a mesocosm experiment in a cold, coastal surface environment and a subtropical
oligotrophic environment of the Northern Atlantic Gyre. As mentioned above, the following comparisons concerning specificity of the primers are based on the program ROSE of the TOOLKIT
software package, though the specificity of the primers when compared using the PROBE MATCH function within the RDP database are also provided (Table 1).
The oligonucleotides designed to specifically amplify the Alphaproteobacteria were identical or similar (Alf28f and Alf684r, respectively) to those suggested for use as molecular probes by
Ashelford et al. (2002), and in one case (Alf684r; Table 3) similar to a primer proposed by Blackwood et al. (2005). The primer sequence of reverse primer Alf684r was homologous to that of a
relatively large number (242) of bacteria outside the Alphaproteobacteria, mainly Fusobacteria and bacteria belonging to the orders Desulfovibrionales, Desulfobacterales and
Desulfuromonadales of the Deltaproteobacteria. In contrast, the sequence of forward primer Alf28f was homologous only to 13 16S rRNA gene sequences outside the target group, equally
distributed among the Gammaproteobacteria, Deltaproteobacteria and the Verrucomicrobia. Due to the high specificity of the forward primer and the high AT, the application of both primers as
a primer pair in a PCR resulted in the amplification of 16S rRNA gene fragments from organisms that all belonged to the target group (Table 4). This primer pair therefore demonstrated the
advantage of both primers of a primer pair being biased towards a particular bacterial group of interest, rather than one group-specific primer combined with a second, Bacteria or universal
16S rRNA gene primer.
Most of the sequences that were detected in either of the two clone libraries belong to members of the Roseobacter group and the genus Sphingomonas, but several were also members of the
genera Rhodobium and Brucella. However, the rest of the sequences belonged to a wide range of different groups of the Alphaproteobacteria (Supplementary Table S1). This demonstrates the
value of the group-specific primer approach in detecting specifically a wide genetic diversity within each taxonomic group.
Ashelford et al. (2002) also suggested a number of oligonucleotides specific for the Betaproteobacteria. Our independent search led us to two primers (Beta359f, Beta682r; Table 1), which
were identical to two of those suggested by Ashelford et al. (2002). Reverse primer Beta682r exploits the same discriminatory sequence stretch as primer Beta680F used by Blackwood et al.
(2005) as a forward primer, although it is different in length and precise sequence position (Table 3). All sequences analysed from the mesocosm sample were from Betaproteobacteria (Table
4). In contrast, despite the high specificity of the primers, only one-third of the sequences from the Northern Atlantic Gyre clone library were from Betaproteobacteria (Table 4).
Interestingly, three of the clones detected in the clone libraries from the Northern Atlantic Gyre show sequence similarities (at 92–95% sequence similarity) to 16S rRNA gene sequences of
Burkholderia (Supplementary Table S1)—also prevalent in the Sargasso Sea data set (Venter et al., 2004).
To test whether this low yield of positive hits could be improved using a nested reamplification-PCR approach, we also prepared and screened a second clone library prepared using a two-step
nested PCR approach. This used an aliquot of the PCR product obtained with primers 9bfm/1512uR as template for nested reamplification with primers Beta359f/682r in a second PCR. Again, only
about one in four of the 25 sequences screened from this clone library were from Betaproteobacteria. The reason for this low yield of positive hits from this oligotrophic environment is not
known. We believe that the use of primer Beta682r generally results in the group-specific amplification of 16S rRNA gene fragments as the last 3′-end (G) base of the primer is highly
specific for the vast majority of Betaproteobacteria sequences in the ROSE database. This is supported by the fact that the clone libraries prepared from the coastal sample proved to be
composed entirely of target sequences (Table 4). However, the low specificity at the 3′-end of primers Beta359f (most 16S rRNA gene sequences have three guanosines at E. coli positions
376–378), combined with the potentially low abundance of Betaproteobacteria in the Northern Atlantic Gyre sample may have led to the low percentage of target hits in this clone library
(Table 4).
As in the case of the Alphaproteobacteria, one of the primers (Gamma359f) appears to have a high number (203) of matches outside the target group (Table 1). However, more than 160 of these
sequence hits are due to a large number of sequences for a small number (five) of particular bacterial species. For example, 45 non-target hits are due to homologous sequences within the 16S
rRNA gene of Acetobacter, Gluconobacter and Gluconacetobacter. Furthermore, 118 hits are due to the Ralstonia (54) and Burkholderia (64) sequence hits. The reason for the high number of
sequences obtained from these bacteria in the databases is because they are important pathogens. Given this, and the higher specificity of the reverse primer Gamma871r (Table 1), it is not
surprising that the specificity found in both clone libraries is 100% (Table 4). This is further confirmed by the fact that the application of the Betaproteobacteria-specific primers
demonstrated the presence of sequences with 92–95% sequence similarity to 16S rRNA gene sequences of Burkholderia as the next nearest isolated species (Supplementary Table S1), but no such
sequences were detected within the clone libraries produced with the Gammaproteobacteria-specific primers (Supplementary Table S1).
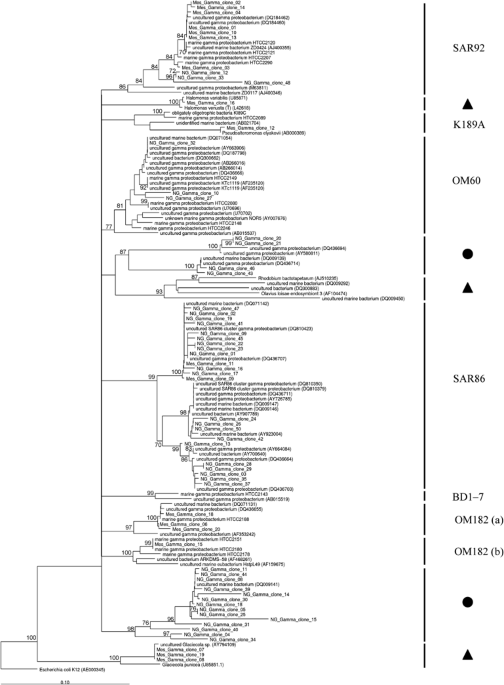
Many of the sequences obtained from the mesocosm sample have a high sequence similarity to 16S rRNA gene sequences that fall within phylogenetic clades that belong to the oligotrophic marine
Gammaproteobacteria (OMG) group (Figure 1; Supplementary Table S1). This group of Gammaproteobacteria was introduced by Cho and Giovannoni (2004) to indicate that all isolates were able to
grow only in low-nutrient (oligotrophic) media. Phylogenetic analysis of their 16S rRNA gene sequences further confirmed that they form independent phylogenetic clades, which together
comprise the OMG group of Gammaproteobacteria (Cho and Giovannoni, 2004). The fact that Gammaproteobacteria 16S rRNA gene sequences from the mesocosm (Figure 1) cluster within two (SAR92 and
OM182) of the five OMG clades identified by Cho and Giovannoni (2004) may indicate that this phylogenetic clade represents a group within the Bacteria that is genetically diverse and occurs
also in coastal nutrient-rich environments. In fact, Stingl et al. (2007) state for the SAR92 clade, into which several of the 16S rRNA gene sequences from the mesocosm sample cluster, that
‘the peak of abundance correlates with the relatively high nutrient concentrations found in an upwelling region off the Oregon coast. In the lower nutrient regions farther off the coast,
the abundance of the SAR92 was low, close to the limit of detection’. A possible explanation for this difference may be that under laboratory conditions, SAR92 isolates grow only under
low-nutrient conditions while in their natural environment they thrive at higher nutrient concentrations such as provided by coastal environments. Alternatively, the phylotypes that we
detected in the mesocosm samples may be physiologically different from those isolated by Cho and Giovannoni (2004).
Phylogenetic analysis of representative 16S rRNA gene nucleotide sequences from the Gammaproteobacteria clone libraries prepared from samples from mesocosm (coastal) and Northern Atlantic
Gyre (open ocean) samples and representative sequences from the NCBI and ARB sequence database. The tree was calculated from a nucleotide alignment of 16S rRNA gene fragments (356 bases)
using the neighbour-joining method within ARB, with Jukes–Cantor corrections and a maximum frequency filter (Ludwig et al., 2004). Escherichia coli (accession J01859) was used as outgroup.
The confidence of branch points was determined by three separate analyses (maximum likelihood, neighbour-joining, maximum parsimony), with multifurcations indicating branch points that were
collapsed using a strict consensus rule until supported in all three analyses. Values of 100 bootstrap replicates (calculated using the neighbour-joining method) are given as numbers at
branching points, but those