Producing gm-csf: a unique t helper subset?
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
GM-CSF-producing helper T cells have previously been identified to serve a nonredundant function in the initiation of autoimmune inflammation. An article by Sheng _et al_. recently published
by _Cell Research_ now suggests that the differentiation program of GM-CSF-producing cells from naïve CD4+ T cells is distinct from that of Th1 and Th17 cells, and is regulated by the
IL-7-STAT5 axis. Interferon-γ (IFN-γ)-producing T helper type 1 (Th1) cells and interleukin 17 (IL-17)-producing T helper (Th17) cells have been shown to drive chronic inflammatory diseases.
However, in the animal model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE), IFN-γ, the Th1-inducing cytokine IL-12 and Th17-associated cytokines such as IL-17A,
IL-17F, IL-21 and IL-22 have all been shown to be dispensable for the development of EAE. It was not until 2011, when the pathogenicity of Th17 cells has been associated with their
production of granulocyte-macrophage colony-stimulating factor (GM-CSF)1,2. GM-CSF is a potent proinflammatory cytokine that is responsible for the recruitment, maturation and activation of
innate immune cells. In this regard, GM-CSF production by T cells has been associated with several autoimmune diseases, such as multiple sclerosis, rheumatoid arthritis and myocarditis. The
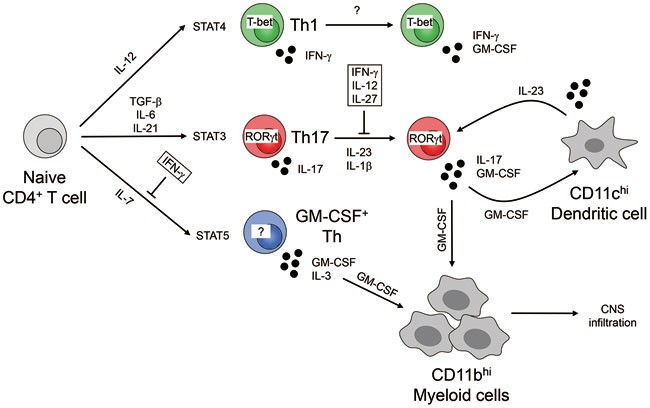
groups of Becher and Rostami demonstrated that IL-1- and IL-23-induced production of GM-CSF by CNS-infiltrating CD4+ T cells is essential for the induction of EAE1,2. These studies also
demonstrated that IL-1 and IL-23 drive the differentiation of Th1 and Th17 effector cells into highly pathogenic GM-CSF-producing CD4+ T cells (Figure 1). GM-CSF thereby exerts a
nonredundant function in EAE pathogenicity, regardless of the polarization pattern of CD4+ effector T cells. Although IL-23 promotes GM-CSF production in Th17 cells, the two studies have
discordant results about the role of RORγt in driving GM-CSF production. The key questions that have not yet been answered by these studies are, whether naïve CD4+ T cells can directly
differentiate into GM-CSF-producing effector CD4+ T cells, whether these effector CD4+ T cells represent a unique T helper cell lineage and which signaling pathways might regulate this T
cell differentiation. In a recent paper published in _Cell Research_, Sheng _et al_.3 demonstrate that conditional _Stat5_-knockout mice (_Stat5_flox/flox _Cd4-Cre_+ mice) are resistant to
EAE development. Although, the role of STAT5 in T cell-mediated autoimmune diseases has not been well characterized, this result is interesting and surprising since STAT5-mediated signaling
is essential for regulatory T cell (Treg) development and inhibits the differentiation of Th17 cells4,5. To determine whether _Stat5_-deficient CD4+ T cells have an intrinsic
encephalitogenic defect, _Stat5_−/− CD4+ T cells were transferred into _Rag2_−/− mice and EAE was induced by immunization with MOG35-55 and Complete Freund's Adjuvant (CFA). _Rag2_−/−
mice reconstituted with _Stat5_−/− CD4+ T cells had significantly reduced EAE disease incidence and severity. Although there were fewer CNS-infiltrating CD4+ T cells at the peak of the
disease, the resistance to EAE was not due to decreased homing to or survival in the CNS. The percentage of Th1 and Th17 cells among CNS-infiltrating CD4+ T cells was also comparable between
_Rag2_−/− mice that were reconstituted with _Stat5_+/+ or _Stat5_−/− CD4+ T cells. However, MOG35-55-specific _Stat5_−/− CD4+ T cells produced significantly less GM-CSF. Further experiments
showed that IL-7-mediated STAT5 signaling promoted the expression of GM-CSF in both naïve and effector CD4+ T cells, and antibody-mediated blockage of IL-7 signaling resulted in decreased
GM-CSF expression in CNS-infiltrating CD4+ T cells. Chromatin immunoprecipitation (ChIP) analysis showed that IL-7-activated STAT5 directly bound to promoter regions of the _Csf2_ gene,
which encodes GM-CSF. These results are in agreement with a recent study showing that STAT5-mediated signaling induced GM-CSF expression in human naïve and memory CD4+ T cells, whereas STAT3
signaling blocked it6. In fact, dysregulation of the IL-7/IL-7R axis has long been implicated in autoimmune diseases, such as type 1 diabetes, multiple sclerosis and rheumatoid
arthritis7,8. Thus, the results from Sheng _et al_. may now provide a mechanistic link between IL-7/STAT5-mediated signaling and T helper cell-mediated pathogenicity. To characterize whether
GM-CSF-producing CD4+ T cells represent a unique T helper cell lineage, Sheng _et al_. performed microarray analysis and identified 211 genes that were expressed in GM-CSF+ Th cells, but
not in Th1, Th17 or naïve CD4+ T cells. As GM-CSF-producing CD4+ T cells did not express the transcription factors RORγt and T-bet, Sheng _et al_. hypothesize that GM-CSF+ Th cells may
represent a unique Th cell lineage. This is an intriguing hypothesis that will require further investigation. In particular, which transcription factor regulates GM-CSF-producing Th cells,
how plastic are GM-CSF+ Th cells _in vitro_ and _in vivo_, and are other STAT5-inducing cytokines or growth factors (e.g., thymic stromal lymphopoietin (TSLP), IL-3, IL-5, IL-9, IL-15,
GM-CSF, growth hormones, _etc_.) involved in the development of GM-CSF+ Th cells9? REFERENCES * Codarri L, Gyulveszi G, Tosevski V, _et al_. _Nat Immunol_ 2011; 12:560–567. * El-Behi M,
Ciric B, Dai H, _et al_. _Nat Immunol_ 2011; 12:568–575. * Sheng W, Yang F, Zhou Y, _et al_. _Cell Res_ 2014; 24:1387–1402. * Malek TR, Yu A, Vincek V, _et al_. _Immunity_ 2002; 17:167–178.
* Laurence A, Tato CM, Davidson TS, _et al_. _Immunity_ 2007; 26:371–381. * Noster R, Riedel R, Mashreghi MF, _et al_. _Sci Transl Med_ 2014; 6:241ra280. * Ribeiro D, Melao A, Barata JT .
_Adv Biol Regul_ 2013; 53:211–222. * Lee LF, Logronio K, Tu GH, _et al_. _Proc Natl Acad Sci USA_ 2012; 109:12674–12679. * O'Shea JJ, Paul WE . _Science_ 2010; 327:1098–1102. Download
references AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Immunobiology, Yale University School of Medicine, New Haven, 06520, CT, USA Dietmar Herndler-Brandstetter &
Richard A Flavell * Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, 06520, CT, USA Richard A Flavell Authors * Dietmar Herndler-Brandstetter View author
publications You can also search for this author inPubMed Google Scholar * Richard A Flavell View author publications You can also search for this author inPubMed Google Scholar
CORRESPONDING AUTHOR Correspondence to Richard A Flavell. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Herndler-Brandstetter, D., Flavell, R.
Producing GM-CSF: a unique T helper subset?. _Cell Res_ 24, 1379–1380 (2014). https://doi.org/10.1038/cr.2014.155 Download citation * Published: 21 November 2014 * Issue Date: December 2014
* DOI: https://doi.org/10.1038/cr.2014.155 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative