Jnk modulates foxo3a for the expression of the mitochondrial death and mitophagy marker bnip3 in pathological hypertrophy and in heart failure
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Bcl-2 E1B 19-KDa interacting protein 3 (BNIP3) is a mitochondrial death and mitophagy marker, which is involved in inducing cardiac remodeling post myocardial infarction. In this
study, we show that BNIP3 expression increases in stressed cardiomyocytes _in vitro_ and in response to pressure overload _in vivo,_ and that its transcription is directly related to JNK
activity. BNIP3 expression gradually increased in the first weeks after pressure overload and peaked at the heart failure stage. Ultrastructurally, the mitochondrial area was inversely
proportional to BNIP3 expression. Both JNK and AKT activities increased with pressure overload; however, JNK signaling dominated over AKT signaling for the activation of the transcription
factor FOXO3a and for the transcription of its effector, BNIP3. 3-methyladenine attenuated JNK signaling and significantly decreased BNIP3 expression and reversed cardiac remodeling in heart
failure. Ultrastructurally, the mitochondrial area was significantly increased in the 3-methyladenine group compared with placebo. Moreover, adenoviral gene delivery of dominant negative
JNK in a rat model of pressure overload hypertrophy abolished the increase in BNIP3 expression in response to pressure overload. These results suggest that JNK signaling is a critical
modulator of the transcription factor FOXO3a driving the expression of its effector, BNIP3, in heart failure and that JNK, through BNIP3, induces mitochondrial apoptosis and mitophagy.
SIMILAR CONTENT BEING VIEWED BY OTHERS KLF9 IS ESSENTIAL FOR CARDIAC MITOCHONDRIAL HOMEOSTASIS Article 08 November 2024 REGULATION OF CARDIOMYOCYTE DNA DAMAGE AND CELL DEATH BY THE TYPE 2A
PROTEIN PHOSPHATASE REGULATORY PROTEIN ALPHA4 Article Open access 18 March 2021 CTRP3 ALLEVIATES MITOCHONDRIAL DYSFUNCTION AND OXIDATIVE STRESS INJURY IN PATHOLOGICAL CARDIAC HYPERTROPHY BY
ACTIVATING UPRMT VIA THE SIRT1/ATF5 AXIS Article Open access 26 January 2024 MAIN Heart failure is a clinical syndrome characterized by the activation of the neurohormonal and renin
angiotensin aldosterone system followed by remodeling of the left ventricle (LV) and alterations in the LV geometry.1 The integrity of the endoplasmic reticulum (ER) and the juxtaposed
mitochondria is pivotal for the proper function of the cardiomyocyte. Ultrastructurally, these two organelles are located at very close proximity and crosstalk with each other via calcium
signaling.2, 3, 4, 5, 6 In heart failure, both the ER and the mitochondria, and each on its own, execute death signals that take the form of programmed apoptotic and autophagic cell death.7
The decline in cardiac function of heart failure patients is in part due to the loss of the diseased cardiac myocytes in the form of necrotic, apoptotic and autophagic cell death. The Bcl-2
family proteins serve as a critical death regulators that reside immediately upstream of the mitochondria. They consist of anti-apoptotic members such as Bcl-2 protein and pro-apoptotic
members. The pro-apoptotic Bcl-2 members are subdivided into ‘multidomain’ and ‘Bcl-2 Homology (BH3)’ proteins. Multidomain pro-apoptotic proteins such as Bax and Bak display sequence
conservation in the BH domains 1–3 and their expression is directly regulated by the anti-apoptotic Bcl-2 protein. On the other hand, the BH3-only members display sequence conservation only
in the _α_-helical BH3 region, which constitutes the critical death domain.8 Of the BH3 members, the Bcl-2 E1B 19-KDa interacting protein 3 (BNIP3) is unique in the sense that it induces
mitochondrial apoptosis as well as mitochondrial autophagy (mitophagy).9 In the initial phase of apoptosis, BNIP3 inserts into the outer mitochondrial membrane with the N terminus oriented
into the cytoplasm and the C terminus inside the mitochondria. It induces mitochondria-mediated apoptosis and fragmentation by driving mitochondrial permeability transition pore opening,
cytochrome C release and the destruction of the mitochondrial cristae.10, 11 On the other hand, BNIP3 is an autophagy receptor that activates mitophagy in a non-canonical order leading to
their sequestration and subsequent removal.9, 12, 13 What makes BNIP3 more interesting is that, unlike the other Bcl-2 members, it is the effector of the transcription factor FOXO3a in
post-mitotic skeletal muscles and cardiomyocytes.14 Many studies have suggested that BNIP3 expression increases under ischemic condition in cardiac myocytes and that cardiac remodeling is
directly related to BNIP3 expression.15, 16, 17, 18, 19 In this study, we show that BNIP3 is expressed in response to cardiomyocyte stressors, such as phenylephrine (PE) or calcium, _in
vitro_ and to pressure overload _in vivo_. Moreover, we show how the interplay between AKT and JNK signaling modulates FOXO3a for the transcription of its effector BNIP3. Moreover, we show
that 3-methyladenine (3 MA), by interfering with JNK signaling, modulates the expression of the mitochondrial death and mitophagy marker BNIP3 _in vivo_, and reverses cardiac remodeling in a
rat model of pressure overload-induced heart failure. This signaling pathway was further validated via the adenoviral gene delivery of dominant negative JNK (Ad-DN-JNK) in a rat model of
pressure overload hypertrophy (POH). RESULTS BNIP3 IS THE EFFECTOR OF FOXO3A AND ITS EXPRESSION IS UPREGULATED IN PE-STRESSED CARDIOMYOCYTES. 3 MA INHIBITED THE INCREASE IN BNIP3 EXPRESSION
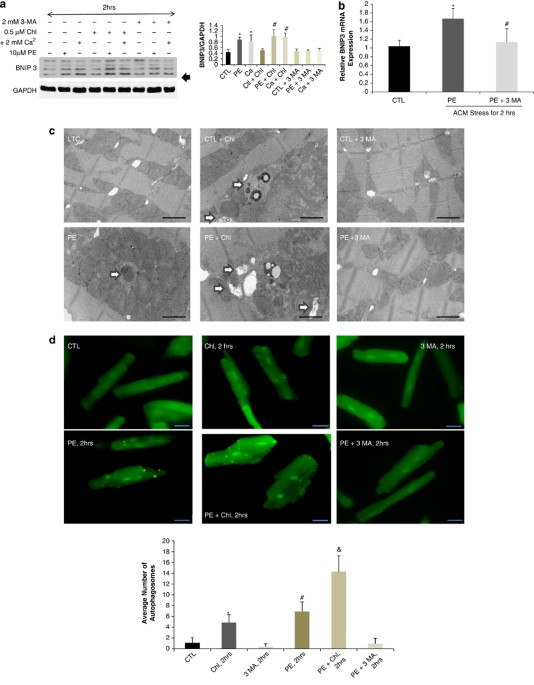
IN PE-STRESSED CARDIAC MYOCYTES _IN VITRO_ BNIP3 expression is increased by two-fold 2 h after cardiomyocyte stress with PE or calcium _in vitro_. BNIP3 expression was also increased with
the addition of chloroquine, a well-known autophagolysosome fusion inhibitor, in stressed cardiomyocytes, whereas 3 MA, an autophagy induction inhibitor, inhibited the increase in BNIP3
expression Figure 1a. The relative BNIP3 mRNA expression was significantly increased in PE-stressed cardiomyocytes for 2 h and its increase was significantly inhibited by 3 MA Figure 1b.
Autophagosomes, containing mitochondria with different stages of vacuolar degeneration, were observed in chloroquine, PE and PE plus chloroquine-stressed cardiomyocytes Figure 1c and
Supplementary Figure 1. The number of autophagosomes increased with the addition of chloroquine and in PE-stressed cardiomyocytes for 2 h with the highest number of autophagosomes observed
in the PE plus chloroquine-treated cardiomyocytes. 3 MA inhibited the formation of autophagosomes in PE-stressed cardiomyocytes Figure 1d. This suggests that the increase in autophagosomes
with PE treatment is due to the increase in autophagosomes formation rather than the consequence of a degradation removal problem. The overexpression of FOXO3a using an adenovirus containing
constitutively active FOXO3a (Ad-FOXO3a) increased the expression of BNIP3 in cardiac myocytes _in vitro_ compared with adenovirus green fluorescent protein (Ad-GFP) and dominant negative
FOXO3a (Ad-DN-FOXO3a), respectively Figure 1e. Moreover, the overexpression of BNIP3 in eGFP-LC3 expressing cardiomyocytes by simultaneous infection with an adenovirus containing BNIP3
(Ad-BNIP3) and another with eGFP-LC3 (Ad-eGFP-LC3), robustly increased the number of autophagosomes compared with adenovirus Null (Ad-Null) and adenovirus Sh BNIP3 (Ad-Sh BNIP3)-infected
cardiomyocytes, respectively Figure 1f. Western blot data shown in Supplementary Figure 2. Ultrastructurally, BNIP3 overexpression in cultured cardiac myocytes was associated with a marked
increase in autophagosomes and robust decrease in mitochondrial area compared with Ad-Null and Ad-Sh BNIP3-infected cardiac myocytes, respectively Figure 1g and Supplementary Figure 3. 3 MA
REVERSED CARDIAC REMODELING AND IMPROVED CARDIAC FUNCTION IN HEART FAILURE 3 MA treatment reversed cardiac remodeling in heart failure compared with the placebo group as shown in Figure 2a.
Transthoracic echocardiography data are shown in the Supplementary Table 1. There was no difference in the heart weight _versus_ the body weight in both the groups. There was no difference
in LV septal (LVSd) and posterior wall (LVPWd) thickness between the 3 MA and the placebo groups. However, there were significant decreases in the LV end diastolic diameter (LVIDd), LV end
diastolic volume (LVEDV), LV end systolic diameter (LVIDs) and LV end systolic volume (LVESV) in the 3 MA group compared with the placebo group, respectively Figure 2b. This was accompanied
by significant increases in LV fractional shortening and LV ejection fraction in the 3 MA group compared with the placebo group Figure 2c. Hemodynamic data are shown in the Supplementary
Table 2. There was a trend in improved LV contractility and efficiency at baseline as determined by pressure–volume loop measurements in the 3 MA group compared with the placebo group, but
with a statistically significant shift of V0 to the left in the 3 MA group compared with placebo Figure 2d and Supplementary Table 2. The 3 MA group had significantly lower end diastolic
pressure, tau and end-diatloic pressure-volume relationships (EDPVR) compared with the placebo group suggesting improved relaxation of the LV. Moreover, The 3 MA group significantly
increased their LV contractility with _β_-adrenergic stimulation in response to escalating doses of dobutamine infusion compared with the placebo group Figure 2e. 3 MA SIGNIFICANTLY
DECREASED THE EXPRESSION OF FOXO3A EFFECTOR, BNIP3 AND MURF-1, AND RESTORED MITOCHONDRIAL CRISTAE IN HEART FAILURE The expression of FOXO3a effectors, the mitophagy marker BNIP3 and the
atrophy marker MurF-1, are significantly upregulated in heart failure compared with age-matched controls. 3 MA significantly attenuated the expression of these markers in heart failure
compared with the placebo group Figure 2f. Ultrastructurally, the placebo group had tortuous-looking myofibers with myofibrillar disarray and an increase in the autophagolysosomes. Also,
there was dilatation of the junctional sarcoplasmic reticulum (JSR) and T-tubules, most of them being occupied with lamellar structures. The intermyofibrillar mitochondria (IMFM) were
fragmented with severe loss in their area and hazy-looking cristae compared with age-matched control. 3 MA treatment reversed the tortuousity of the myofibers and the myofibrillar disarray,
but did not improve JSR dilatation. 3 MA treatment significantly decreased IMFM fragmentation as measured by the decrease in the average area per mitochondrion and restored their cristae
Figures 2g and h. Figure 2i show the correlation between BNIP3 expression and the average area per mitochondrion. The decrease in BNIP3 expression by 3 MA resulted in significant increase in
the average area per mitochondrion. PROLONGED JNK ACTIVATION OVERRIDES THE INHIBITORY EFFECT OF AKT ON FOXO3A AND ACTIVATES THE LATTER FOR THE EXPRESSION OF ITS EFFECTORS, BNIP 3 AND MURF-1
JNK and AKT have opposite effects on FOXO3a. JNK activates FOXO3a whereas AKT inhibits its activity. Moreover, it has been shown that JNK potentiates AKT activity via its phosphorylation at
Thr450 and increases the interaction of AKT with its upstream effectors, PI3K1 and PDK.20 Phosphorylated JNK and AKT are significantly increased in heart failure compared with age-matched
control. However, and despite the increases in AKT activity and the phosphorylation of FOXO3a at Ser253, there were significant increases in FOXO3a effectors, BNIP3 and MurF-1 in the HF
Placebo group. The long-term administration of 3 MA _in vivo_ attenuated JNK activity and significantly decreased the expression of FOXO3a effectors despite the decreases in AKT activity and
the decrease in FOXO3a phosphorylation by AKT Figure 3a. From the above, we hypothesized that the long-term activation of JNK opposes the inhibitory effect of AKT on FOXO3a, via its
phosphorylation at sites that are yet to be identified, and thus override the inhibitory effect of AKT on FOXO3a for the activation of the latter, promoting the transcription of FOXO3a
effectors, BNIP3 and MurF-1, respectively. In other words, the phosphorylation of FOXO3a by AKT is dependent on JNK activity and that prolonged JNK signaling dominate over AKT signaling and
promotes or activates FOXO3a, despite its phosphorylation by AKT, for the transcription and the expression of its effectors BNIP3 and MurF-1, respectively. Moreover, prolonged JNK activity
phosphorylates Bcl-2 and leads to the increase in Bax to Bcl-2 ratio Figure 3b. 3 MA did not affect ERK or P38 activity _in vivo_ Figure 3b. In order to prove the above hypothesis, we used
an Ad-DN-JNK that was delivered via a cross-clamp technique followed by ascending aortic banding (AAB). Saline and an Ad-GFP were used as controls. Transthoracic echocardiography data and
M-mode images are shown in the Supplementary Table 3 and Figure 4a, respectively. There were significant decreases in the heart weight to body weight ratio as well as in the septal and
posterior wall thickness and the LV end systolic diameter and volume of the Ad-DN-JNK group compared with the Saline and the Ad-GFP groups, Figures 4b–d. This reflected significant increases
in LV fractional shortening and ejection fraction, Figure 4e. On the molecular level, the decrease in JNK activity using Ad-DN-JNK, prevented the significant increase in AKT activity and
the increase in FOXO3a phosphorylation by AKT in response to pressure overload as shown in the Saline and the Ad-GFP groups, Figure 4f. Moreover, the continuous decrease in JNK activity, for
2 weeks, inhibited the increase in BNIP 3 expression, despite the decrease in AKT activity, and inhibited the increases in LC3-2 to LC3-1 ratio and Bax to Bcl-2 ratio in response to
pressure overload as shown in the Saline and Ad-GFP groups, respectively Figure 4f. FOXO3A EFFECTORS, BNIP3 AND MURF-1, ARE SIGNIFICANTLY UPREGULATED IN POH AND PEAK IN HEART FAILURE In
order to understand the timing at which this signaling pathway is altered in response to pressure overload, we conducted a kinetic study and examined the expression of the key components in
the pathway at 1 week and 3 weeks of POH and in heart failure. Echo data are shown in the Supplementary Table 4. Our results suggest that JNK and AKT activities are significantly upregulated
1 week after pressure overload and continue to be activated up to heart failure Figure 5a. There were gradual increases in the expression of FOXO3a effectors, BNIP3 and MurF-1, that became
significant 2–3 weeks after pressure overload and peaked in heart failure, despite the increases in FOXO3a phosphorylation at Ser253 by AKT Figure 5a. These results further validate that the
continuous and prolonged JNK activation in response to pressure overload, dominates over AKT signaling and overrides the inhibitory effect of AKT on FOXO3a for the gradual increases in the
expression of FOXO3a effectors, BNIP3 and MurF-1, which peak in heart failure. Moreover, the increases in JNK activity significantly increased Bax to Bcl-2 ratio the first week after
pressure overload and continued to be elevated up to heart failure Figure 5a. This signaling pathway has been validated in human samples of heart failure. There are robust increases in JNK
and AKT activities as well as significant increase in BNIP3 expression, despite the increase in AKT activity, in human samples of HF Figure 5b. Ultrastructurally, there was an increase in
autophagosomes the first week after POH, which continued to be present up to heart failure Figure 6a. There was a significant decrease in the average area per mitochondrion the first week
after POH. The mitochondrial area declined further 3 weeks after pressure overload and was the lowest in heart failure Figures 6a and b. The gradual and significant increase in BNIP3
expression in POH up to heart failure reflected a gradual and significant decrease in the average area per mitochondrion, which was the lowest in heart failure where BNIP3 expression was the
highest Figure 6c. DISCUSSION BNIP3 is a BH3 protein that promotes mitochondrial destruction and signals mitochondrial death via the opening of mitochochondrial permeability transition pore
and the subsequent release of cytochrome C.11 Moreover, it is an autophagy receptor that activates mitophagy in a non-canonical order via the binding and the lipidation of LC3.12 Our data
suggest that BNIP3 expression is significantly upregulated 2 h after cardiomyocyte stress _in vitro_ with PE or calcium. Also, the addition of chloroquine to the cultured medium increased
BNIP3 expression, whereas 3 MA attenuated its expression in stressed cardiomyocytes. The overexpression of BNIP3 in cardiac myocytes, using Ad-BNIP3, robustly increased autophagosomes
formation and significantly decreased mitochondrial area. In cardiac myocytes and in skeletal muscles, BNIP3 is the effector of the transcription factor FOXO3a.14, 21 The activation of
FOXO3a or its inhibition is dependent on its post-translational modification status.22 AKT phosphorylates FOXO3a at Thr32, Ser253 and Ser315 leading to its inhibition, whereas JNK interacts
and phosphorylates FOXO3a, at sites that are still unknown, leading to its activation.23 In heart failure, FOXO3a effectors, BNIP3 and MurF-1 are highly expressed compared with age-matched
controls. The increase in BNIP3 expression leads to increase in mitochondrial destruction, mitochondrial apoptosis and mitophagy; whereas, the increase in MurF-1 and atrogin-1 promote
cardiac atrophy.24, 25, 26 3 MA treatment robustly decreased the expression of FOXO3a effectors, BNIP3 and MurF-1, respectively, and hence attenuated mitochondrial damage and cardiac atrophy
by interfering with JNK signaling. In our model of heart failure, both JNK and AKT were activated; however, AKT failed to inhibit FOXO3a. On the other hand, 3 MA by attenuating JNK activity
reversed this signaling pathway and decreased the expression of FOXO3a effectors and pro-apoptotic marker Bax and, hence, decreased Bax to Bcl-2 ratio. These results led us to hypothesize
that prolonged JNK signaling dominates over AKT and overrides the inhibitory effect of AKT on FOXO3a and forces the latter to re-translocate into the nucleus for the transcription of its
effectors. Moreover, prolonged JNK signaling phosphorylates Bcl-2 and increases the expression of the pro-apoptotic marker Bax, thus increasing Bax over Bcl-2 ratio and cardiomyocyte loss
through apoptosis. This hypothesis was further validated using an adenovirus expressing dominant negative JNK that was delivered _in vivo_ using a cross-clamp technique followed by aortic
constriction for the creation of pressure overload. The continuous inhibition of JNK activation for 2 weeks prevented cardiac hypertrophy and prevented the decline in cardiac function in
response to pressure overload. These results are consistent with those of Choukroun _et al._27, 28 who have shown that the inhibition of JNK suppresses cardiac hypertrophy _in vitro_ and _in
vivo_ in response to endothelin and pressure overload, respectively. On the molecular level, continuous JNK inhibition for 2 weeks by Ad-DN-JNK prevented the increase in AKT activity, BNIP3
expression and the increase in Bax to Bcl-2 ratio in response to pressure overload Figure 7. Our kinetic study shows that JNK and AKT signaling pathways are activated early in the first
week of POH and remains active throughout compensated hypertrophy up to heart failure. However, what is more interesting is that the prolonged and persistent JNK signaling leads to the
gradual but significant rise in FOXO3a transcription factors, which peak in heart failure despite the increase in AKT activity and the increase in FOXO3a phosphorylation at Ser253 by AKT.
This signaling pathway was further validated in human samples of heart failure. The steady and progressive increase in BNIP3 expression leads to progressive and steady increase in
mitochondrial fragmentation and to the gradual decrease in the average area per unit mitochondrion, which is the lowest in heart failure. On the other hand, the attenuation of BNIP3
expression by 3 MA decreased mitochondrial fragmentation and increased the average area per mitochondrion. CONCLUSION BNIP3 induces mitochondrial autophagy and apoptosis early on in POH and
is further increased in heart failure. JNK is a critical regulator of BNIP3 expression _in vivo_. Prolonged JNK signaling dominates and overrides AKT signaling and activates FOXO3a
contributing to the gradual increase in BNIP3 expression in compensated hypertrophy, which peaks in heart failure. The gradual increase in BNIP3 expression causes destruction of the
mitochondria and promotes mitochondrial-induced apoptosis and mitophagy leading to the gradual decline in cardiac function and LV remodeling. 3 MA modulates the expression of BNIP3, _in
vivo_, via the interference with JNK signaling and reverses cardiac remodeling in heart failure. MATERIALS AND METHODS ISOLATION AND CULTURE OF ADULT RAT CARDIOMYOCYTES AND _IN VITRO_
EXPERIMENTS Adult rat ventricular cardiomyocytes were isolated from male Sprague–Dawley rats weighing (250–350 g) as previously described.29, 30 Cells were stressed for 2 h with 10 _μ_M of
PE or with the addition of 2 mM of calcium to the culture medium. Chloroquine was added at a concentration of 0.5 _μ_M for autophagosome–lysosome fusion inhibition, whereas autophagy was
inhibited by the addition of 2.5 mM of 3 MA to the cultured medium. All reagants were purchased from (Sigma Chemicals, St. Louis, MO, USA). After 2 h, the cells were lysed using RIPA buffer
as mentioned in the western blotting section (_n_=3 for each experiment _in vitro_). For electron microscopy, cells were fixed with 3% glutarldehyde for 24 h at 4 °C. The next day, the cells
were detached using a scraper and were sent for electron microscopy for processing as discussed below in the electron microscopy section. For the Autophagic flux experiments, cells were
infected with adenovirus eGFP-LC3 and images were taken using fluorescent microscope, Olympus 1 × 71. WESTERN BLOTTING 30 ug of proteins were loaded and electrophoresed using SDS-PAGE gels
then transferred to a PVDF membrane. The membrane was blocked for 1 h using blocking solution then was incubated with the primary Ab overnight at 4 °C. The following antibodies were used:
GAPDH (Sigma, 1 : 10000 dilution), LC 3, BNIP3, JNK, AKT, p-AKT Ser473, FOXO3a, FOXO3a Ser253, Bax, Bcl-2, ERK, p-ERK, P38 and p-P38 (Cell Signaling, Danvers, MA, USA, 1 : 1000 dilution),
p-JNK (Promega, Madison, WI, USA, 1 : 2000 dilution), MurF-1 (BDM Biosciences, Franklin Lakes, NJ, USA, 1 : 1000 dilution) and BNIP3 human-specific (Abcam, Cambridge, MA, USA, 1 : 1000
dilution). The second day, after three washing steps with TBS-0.05% Tween-20, the blot was incubated with secondary horseradish peroxidase-conjugated antibody (1 : 10000 dilution) for 45
min. The blot was washed three times with TBS- 0.05% Tween-20, then a supersignal west pico chemiluminescent substrate (Thermo Scientific, Barrington, IL, USA) was used for the detection of
protein bands using the film method. Bands densities were quantified using photoshop program and were normalized to GAPDH to correct for variations in protein loading. QUANTITATIVE RT-PCR
Total RNA was isolated using RNeasy Protect Mini kit (Qiagen, Hilden, Germany). Reverse transcription was performed using High Capacity cDNA Reverse Transcription Kits (Applied Biosystems,
Carlsbad, CA, USA) with random oligo-dT priming as followed by the manufacturer's protocols. PCR was performed using an ABI PRISM Sequence Detector System 7500 (Applied Biosystems) with
SYBR Green (BioRad, Hercules, CA, USA) as fluorescent, and ROX (Takara, Otsu, Japan) as a passive reference dye. The PCR primers used were: BNIP3-F, 5′-AGCATGAATCTGGACGAAGC-3′ and BNIP3-R
5′-AACATTTTCTGGCCGACTTG-3′; 18S-F, 5′-tgcggaaggatcattaacgga-3′ and 18S-R, 5′-agtaggagaggagcgagcgacc-3′ was used as an endogeneous-loading control. HUMAN HEART SAMPLES Left ventricular
samples were obtained from explanted human hearts obtained at the time of cardiac transplantation. Non-failing hearts, used as controls, were obtained from donors who died from neurological
diseases or motor-vehicle accidents and who had normal cardiac function. The three donors had a median age of 62. The heart-failure patients had a median age of 60 and their average ejection
fraction was 20±3%. ELECTRON MICROSCOPY Cells and fractions (1 mm3) from fresh ventricles were pre-fixed in a solution of 3% glutaraldehyde overnight at 4 °C, post-fixed in 1% osmium
tetroxide (OsO4), dehydrated in an ascending series of alcohols, and embedded in epoxy resin. Ultrathin sections were stained with uranylacetate and lead citrate. Samples were viewed under a
transmission electron microscope (HITACHI H-7650, Japan). Images were taken at 1 K, 5 K and 12 K magnification. IMAGE ANALYSIS USING IMAGE J Mitochondrial area analysis was done using
Image-J, a public domain Java image-processing program inspired by NIH. Scale setting and calibration were done using the ‘Set Scale and Calibration Menu,’ and measurement parameters were
selected. The mitochondrial area was summed for each image using 12 000 magnified images and the average mitochondrial area per image was calculated in _μ_m2 (total area/number of
mitochondria) after correction by the magnification factor. PRODUCTION OF RECOMBINANT ADENOVIRUSES Recombinant Ad-GFP was prepared as described previously.31 Briefly, The Ad-Easy Adenoviral
vector system (Stratagene, La Jolla, CA, USA) was used to generate recombinant adenoviruses. Full-length _EGFP_ gene was subcloned into the pShuttle vector (containing the cDNA for enhanced
GFP) under the control of CMV promoter. Viral titers were determined by the plaque assay and the absence of replication-competent adenovirus was confirmed by PCR to assess the wild-type E1
region. A dominant-negative JNK adenovirus (Ad-DN-JNK1) was purchased from Seven Hills Bioreagents (Cincinnati, OH, USA). Ad-DN-FOXO3a and Ad-FOXO3a were purchased from Vector Biolabs
(Philadelphia, PA, USA). Adenovirus Null, BNIP3, Sh BNIP3 and eGFP-LC3 were done at Vector Biolabs. A multiplicity of infection of 100 has been used in all infection experiments _in vitro_
(_n_=3 for each experiment _in vitro_). For the _in vivo_ experiments, the adenovirus was delivered via a cross clamp technique as described below with an infectious dose of 200 pfu/cell.
EXPERIMENTAL MODEL OF AAB, CROSS CLAMP WITH AAB AND STUDY DESIGN All procedures involving the handling of animals were approved by the Animal Care and Use Committee of the Mount Sinai School
of Medicine and adhered with the Guide for the Care and Use of laboratory Animals published by the National Institutes of Health. The aortic banding model was used to generate pressure
overload-induced hypertrophy and heart failure. Sprague–Dawley rats weighing 180–200 g underwent AAB, as previously described in detail.32 For the _in vivo_ Kinetic study, animals were
killed 1–3 weeks after AAB and at heart failure development (_n_=4 at each time point). Before killing, echocardiography was used to assess LV size and function, whereas the clip placement
was verified at the time of killing. For the heart failure experiment, animals that developed heart failure were randomized to receive placebo or 3 MA for one month. 3 MA was administered
intraperitoneally at a dose of 40 mg/kg/day to inhibit autophagy induction _in vivo_. Age-matched sham-operated animals were used as control (_n_=4 in each group). The cross clamp surgery
with gene transfer and AAB was performed as previously described in detail.33, 34 Briefly, the chest was opened at midline between the second and the fifth intercostals space. The aorta and
the pulmonary arteries were cross-clamped simultaneously and the virus (Ad-GFP _versus_ Ad-DN-JNK)/saline was injected into the LV. The cross clamp duration was 45 s and the adenovirus dose
used was 200 PFU/cell. After adenoviral delivery, AAB was performed and animals were studied 2 weeks later (_n_=4 in each group). ECHOCARDIOGRAPHY Transthoracic echocardiography was
performed using a vivid 7 echocardiography apparatus with a 14 MHZ probe (i13L probe, General Electric, New York, NY, USA). Long-axis parasternal views and short-axis parasternal two
dimensional (2D) views, at the mid-papillary level, of the LV were obtained to calculate the LVEDV and LVESV volumes as well as the ejection fraction of the LV. INVASIVE PRESSURE-VOLUME LOOP
MEASUREMENTS OF THE LV Hemodynamics were recorded subsequently through a Scisense P–V Control Unit (FY897B, London, ON, Canada). The intrathoracic inferior vena cava was transiently
occluded to decrease venous return during the recording to obtain load-independent P–V relationships. Linear fits were obtained for end-systolic pressure volume relationships and EDPVR. At
the end of the experiment, 50 _μ_l of 30% NaCl were slowly injected into the external jugular vein for ventricular parallel conductance (Gp) measurement as previously described.35, 36 Blood
resistivity was measured using a special probe (Scisense). Volume measurements were initially obtained as blood conductance and calibrated using the Baan equation,37 and pressure sensors
were calibrated as per manufacturer's instructions. STATISTICAL ANALYSIS Results are shown as mean±S.D. Statistical significance was determined using Student–Newman–Keuls test. A
_P_-value of <0.05 was considered statistically significant. ABBREVIATIONS * ER: endoplasmic reticulum * JSR: junctional sarcoplasmic reticulum * IMFM: intermyofibrillar mitochondria *
mPTP: mitochondrial permeability transition pore * BNIP3: Bcl-2 E1B 19-KDa interacting protein 3 * PE: phenylephrine * 3 MA: 3-methyladenine * Ad-GFP: adenovirus green fluorescent protein *
Ad-Null: adenovirus Null * Ad-DN-JNK: adenovirus dominant negative JNK * Ad-FOXO3a: adenovirus constitutively active FOXO3a * Ad-DN-FOXO3a: adenovirus dominant negative FOXO3a * Ad-BNIP3:
adenovirus BNIP3 * Ad-Sh BNIP3: adenovirus small hairpin BNIP3 * Ad-eGFP-LC3: adenovirus enhanced green fluorescent -light chain 3 protein REFERENCES * Mann DL . Left ventricular size and
shape: determinants of mechanical signal transduction pathways. _Heart Fail Rev_ 2005; 10: 95–100. Article Google Scholar * Giorgi C, De Stefani D, Bononi A, Rizzuto R, Pinton P .
Structural and functional link between the mitochondrial network and the endoplasmic reticulum. _Int J Biochem Cell Biol_ 2009; 41: 1817–1827. Article CAS Google Scholar * Walter L,
Hajnoczky G . Mitochondria and endoplasmic reticulum: the lethal interorganelle cross-talk. _J Bioenerg Biomembr_ 2005; 37: 191–206. Article CAS Google Scholar * Boncompagni S, Rossi AE,
Micaroni M, Beznoussenko GV, Polishchuk RS, Dirksen RT _et al_. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. _Mol Biol
Cell_ 2009; 20: 1058–1067. Article CAS Google Scholar * Griffiths EJ, Rutter GA . Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. _Biochim
Biophys Acta_ 2009; 1787: 1324–1333. Article CAS Google Scholar * Rizzuto R, Pozzan T . Microdomains of intracellular Ca2+: molecular determinants and functional consequences. _Physiol
Rev_ 2006; 86: 369–408. Article CAS Google Scholar * Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A . Life and death partners: apoptosis, autophagy and the cross-talk between them.
_Cell Death Differ_ 2009; 16: 966–975. Article CAS Google Scholar * Gross A, McDonnell JM, Korsmeyer SJ . BCL-2 family members and the mitochondria in apoptosis. _Genes Dev_ 1999; 13:
1899–1911. Article CAS Google Scholar * Zhang J, Ney PA . Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. _Cell Death Differ_ 2009; 16: 939–946. Article CAS Google
Scholar * Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnaune-Pelloquin L . The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by
distinct mechanisms. _EMBO Rep_ 2010; 11: 459–465. Article CAS Google Scholar * Quinsay MN, Lee Y, Rikka S, Sayen MR, Molkentin JD, Gottlieb RA _et al_. Bnip3 mediates permeabilization
of mitochondria and release of cytochrome c via a novel mechanism. _J Mol Cell Cardiol_ 2010; 48: 1146–1156. Article CAS Google Scholar * Quinsay MN, Thomas RL, Lee Y, Gustafsson AB .
Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. _Autophagy_ 2010; 6: 17–24. Article Google Scholar * Glick D, Barth S, Macleod KF .
Autophagy: cellular and molecular mechanisms. _J Pathol_ 2010; 221: 3–12. Article CAS Google Scholar * Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P _et al_.
FoxO3 controls autophagy in skeletal muscle _in vivo_. _Cell Metab_ 2007; 6: 458–471. Article CAS Google Scholar * Metukuri MR, Beer-Stolz D, Namas RA, Dhupar R, Torres A, Loughran PA _et
al_. Expression and subcellular localization of BNIP3 in hypoxic hepatocytes and liver stress. _Am J Physiol Gastrointest Liver Physiol_ 2009; 296: G499–G509. Article CAS Google Scholar
* Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG _et al_. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in
mice. _J Clin Invest_ 2007; 117: 2825–2833. Article CAS Google Scholar * Gang H, Hai Y, Dhingra R, Gordon JW, Yurkova N, Aviv Y _et al_. A novel hypoxia-inducible spliced variant of
mitochondrial death gene bnip3 promotes survival of ventricular myocytes. _Circ Res_ 2011; 108: 1084–1092. Article CAS Google Scholar * Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB
. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. _Am J Physiol Heart Circ Physiol_ 2008; 295: H2025–H2031. Article CAS Google
Scholar * Regula KM, Ens K, Kirshenbaum LA . Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. _Circ Res_ 2002; 91:
226–231. Article CAS Google Scholar * Shao Z, Bhattacharya K, Hsich E, Park L, Walters B, Germann U _et al_. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte
survival after hypoxic injury _in vitro_ and _in vivo_. _Circ Res_ 2006; 98: 111–118. Article CAS Google Scholar * Mammucari C, Schiaffino S, Sandri M . Downstream of Akt: FoxO3 and mTOR
in the regulation of autophagy in skeletal muscle. _Autophagy_ 2008; 4: 524–526. Article CAS Google Scholar * Sandri M . Signaling in muscle atrophy and hypertrophy. _Physiology
(Bethesda)_ 2008; 23: 160–170. CAS Google Scholar * Greer EL, Brunet A . FOXO transcription factors at the interface between longevity and tumor suppression. _Oncogene_ 2005; 24:
7410–7425. Article CAS Google Scholar * Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ _et al_. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by
participating in an SCF ubiquitin ligase complex. _J Clin Invest_ 2004; 114: 1058–1071. Article CAS Google Scholar * Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ _et al_.
Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. _J Clin Invest_ 2007; 117: 3211–3223. Article CAS Google Scholar *
Willis MS, Rojas M, Li L, Selzman CH, Tang RH, Stansfield WE _et al_. Muscle ring finger 1 mediates cardiac atrophy _in vivo_. _Am J Physiol Heart Circ Physiol_ 2009; 296: H997–H1006.
Article CAS Google Scholar * Choukroun G, Hajjar R, Fry S, del Monte F, Haq S, Guerrero JL _et al_. Regulation of cardiac hypertrophy _in vivo_ by the stress-activated protein
kinases/c-Jun NH(2)-terminal kinases. _J Clin Invest_ 1999; 104: 391–398. Article CAS Google Scholar * Choukroun G, Hajjar R, Kyriakis JM, Bonventre JV, Rosenzweig A, Force T . Role of
the stress-activated protein kinases in endothelin-induced cardiomyocyte hypertrophy. _J Clin Invest_ 1998; 102: 1311–1320. Article CAS Google Scholar * Piper HM, Probst I, Schwartz P,
Hutter FJ, Spieckermann PG . Culturing of calcium stable adult cardiac myocytes. _J Mol Cell Cardiol_ 1982; 14: 397–412. Article CAS Google Scholar * Wold LE, Ren J . Mechanical
measurement of contractile function of isolated ventricular myocytes. _Methods Mol Med_ 2007; 139: 263–270. Article Google Scholar * Kim M, Oh JK, Sakata S, Liang I, Park W, Hajjar RJ _et
al_. Role of resistin in cardiac contractility and hypertrophy. _J Mol Cell Cardiol_ 2008; 45: 270–280. Article CAS Google Scholar * Del Monte F, Butler K, Boecker W, Gwathmey JK, Hajjar
RJ . Novel technique of aortic banding followed by gene transfer during hypertrophy and heart failure. _Physiol Genomics_ 2002; 9: 49–56. Article CAS Google Scholar * Hajjar RJ, Schmidt
U, Matsui T, Guerrero JL, Lee KH, Gwathmey JK _et al_. Modulation of ventricular function through gene transfer _in vivo_. _Proc Natl Acad Sci USA_ 1998; 95: 5251–5256. Article CAS Google
Scholar * del Monte F, Hajjar RJ . Efficient viral gene transfer to rodent hearts _in vivo_. _Methods Mol Biol_ 2003; 219: 179–193. CAS PubMed Google Scholar * Pacher P, Nagayama T,
Mukhopadhyay P, Batkai S, Kass DA . Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. _Nat Protoc_ 2008; 3: 1422–1434. Article CAS
Google Scholar * Porterfield JE, Kottam AT, Raghavan K, Escobedo D, Jenkins JT, Larson ER _et al_. Dynamic correction for parallel conductance, GP, and gain factor, alpha, in invasive
murine left ventricular volume measurements. _J Appl Physiol_ 2009; 107: 1693–1703. Article Google Scholar * Baan J, van der Velde ET, de Bruin HG, Smeenk GJ, Koops J, van Dijk AD _et al_.
Continuous measurement of left ventricular volume in animals and humans by conductance catheter. _Circulation_ 1984; 70: 812–823. Article CAS Google Scholar Download references
ACKNOWLEDGEMENTS This work was supported by NIH R01 HL083156, HL080498, HL093183 and P20HL100396 to (RJH) and by a 2011 Research Fellowship Award from the Heart Failure Society of America to
(AHC). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Cardiovascular Institute, Mount Sinai School of Medicine, New York, 10029, NY, USA A H Chaanine, D Jeong, L Liang, E R Chemaly, K Fish
& R J Hajjar * Pathology Department, Mount Sinai School of Medicine, New York, 10029, NY, USA R E Gordon Authors * A H Chaanine View author publications You can also search for this
author inPubMed Google Scholar * D Jeong View author publications You can also search for this author inPubMed Google Scholar * L Liang View author publications You can also search for this
author inPubMed Google Scholar * E R Chemaly View author publications You can also search for this author inPubMed Google Scholar * K Fish View author publications You can also search for
this author inPubMed Google Scholar * R E Gordon View author publications You can also search for this author inPubMed Google Scholar * R J Hajjar View author publications You can also
search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to R J Hajjar. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest.
ADDITIONAL INFORMATION Edited by RA Knight Supplementary Information accompanies the paper on Cell Death and Disease website SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION (DOC 7594 KB)
RIGHTS AND PERMISSIONS This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit
http://creativecommons.org/licenses/by-nc-nd/3.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Chaanine, A., Jeong, D., Liang, L. _et al._ JNK modulates FOXO3a for the
expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. _Cell Death Dis_ 3, e265 (2012). https://doi.org/10.1038/cddis.2012.5
Download citation * Received: 12 December 2011 * Revised: 05 January 2012 * Accepted: 05 January 2012 * Published: 02 February 2012 * Issue Date: February 2012 * DOI:
https://doi.org/10.1038/cddis.2012.5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * heart failure * JNK * FOXO3a * BNIP3 * mitochondrial
apoptosis * mitophagy