RETRACTED ARTICLE: Effect of pantoprazole to enhance activity of docetaxel against human tumour xenografts by inhibiting autophagy
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
Autophagy allows recycling of cellular components and may facilitate cell survival after chemotherapy. Pantoprazole inhibits proton pumps and is reported to inhibit autophagy. Here we
evaluate the effects of pantoprazole to modify cytotoxicity of the anticancer drug docetaxel, and underlying mechanisms.
Effects of docetaxel±pantoprazole were studied against wild-type and autophagy-deficient PC3 cells and against four human xenografts. Effects of pantoprazole on autophagy were evaluated by
quantifying LC3-I, LC3-II and p62 proteins in western blots, and by fluorescent microscopy of cells transfected with RFP-GFP-LC3. The distribution of drug effects and of autophagy was
quantified in tumour sections in relation to blood vessels and hypoxia by immunohistochemistry using γH2AX, cleaved caspase-3, Ki67 and LC3/ p62.
Pantoprazole increased the toxicity of docetaxel in vitro, increased docetaxel-induced expression of γH2AX and cleaved caspase-3, and decreased Ki67 in tumour sections. Pantoprazole
increased growth delay of four human xenografts of low, moderate and high sensitivity to docetaxel, with minimal increase in toxicity. Docetaxel led to increased autophagy throughout tumour
sections. Pantoprazole inhibited autophagy, and effects of pantoprazole were reduced against genetically modified cells with decreased ability to undergo autophagy.
Autophagy is a mechanism of resistance to docetaxel chemotherapy that may be modified by pantoprazole to improve therapeutic index.
Causes of resistance to chemotherapy have focused on molecular changes in individual tumour cells, including expression of drug export pumps such as P-glycoprotein and altered expression of
tubulin isotypes, which convey resistance to taxanes (Bradley and Ling, 1994; Terry et al, 2009; Ploussard et al, 2010). Other mechanisms depend on the solid tumour microenvironment
including problems of limited delivery of anticancer drugs to tumour cells that are distal to functional blood vessels (Lankelma et al, 1999; Tannock et al, 2002; Huxham et al, 2004; Tredan
et al, 2007), and resistance of these slowly proliferating, poorly nourished (often hypoxic) cells to cycle-dependent chemotherapy. We have shown that limited distribution of activity in
poorly nourished or hypoxic regions of solid tumours is common to many anticancer drugs, including docetaxel, and is an important cause of therapeutic resistance (Saggar et al, 2013).
Autophagy is a cellular process of self-consumption characterised by sequestration of bulk cytoplasm, long-lived proteins and cellular organelles into double-membrane vesicles called
autophagosomes, which are delivered to and degraded in lysosomes (White, 2012). Markers of autophagy co-localise in hypoxic and poorly nourished regions of tumours (Hoyer-Hansen and
Jaattela, 2007; Rouschop et al, 2010). Autophagy is prognostic of poor outcome in multiple tumour types (Sivridis et al, 2010; Karpathiou et al, 2011; Sivridis et al, 2011), and high levels
of autophagy have been associated with resistance to systemic therapy in several preclinical and clinical models, presumably because autophagy facilitates survival of stressed or damaged
cells through recycling of cellular breakdown products (Yang et al, 2011). Hence, targeting of autophagy with pharmacological agents may be a mechanism to improve the effectiveness of
anticancer drugs for solid tumours. Current clinical strategies for inhibiting autophagy include the use of hydroxychloroquine (HCQ) and proton pump inhibitors (PPIs) that disrupt lysosomal
pH regulation and thus prevent autolysosome formation and degradation of captured cytoplasmic content. Several clinical trials of HCQ in combination with cytotoxic (docetaxel, temozolomide)
and targeted (gefitinib) agents are underway, based on the premise that inhibition of autophagy by HCQ should enhance the efficacy of these drugs (Poklepovic and Gewirtz, 2014; Rosenfeld et
al, 2014). Inhibitors of autophagy with greater specificity than HCQ are also being developed (McAfee et al, 2012; Deng et al, 2013).
Pantoprazole is a PPI that inhibits the gastric H+, K+-ATPase proton pump; but at higher concentration, PPIs inhibit other proton pumps, including those that acidify endosomes; they have
been reported to inhibit autophagy possibly through inhibiting acidification of endosomes or their fusion with autophagosomes (Udelnow et al, 2011). Proton pump inhibitors have been reported
to sensitise cancer cells and solid tumours to different chemotherapeutic agents (Luciani et al, 2004). Multiple mechanisms are probably involved, but appear to relate to changes in acidity
in intra- and extracellular compartments of tumour cells. Several studies have shown that PPIs such as omeprazole, esomeprazole and pantoprazole have activity against human hematopoietic
and solid tumours; they may revert chemo-resistance in drug-resistant tumours and directly induce killing of tumour cells (Yeo et al, 2004; De Milito et al, 2007; de Milito et al, 2010).
Growing evidence suggests that the major mechanism may be inhibition of autophagy (Levy et al, 2014; Pan et al, 2014; Wang and Wu, 2014; Yang et al, 2014a, 2014b).
In the present study, we report that pantoprazole enhances the in vitro and in vivo activity of docetaxel, a drug in wide clinical use, and provide evidence that the major underlying
mechanism is the inhibition of autophagy.
Human breast carcinoma MCF-7 cells, human vulvar epidermoid carcinoma A-431 cells, and human prostate cancer PC3 and LNCap cells were used. All cell lines were purchased from the American
Type Culture Collection in 2011. MCF-7, A-431 and cells have been maintained in our laboratory and were grown in α-minimum essential medium supplemented with 10% FBS (Hyclone, Logan, UT,
USA). The PC3 and LNCaP cells were grown in Ham’s F-12K medium (Life Technologies Inc., Carlsbad, CA, USA) supplemented with 10% FBS. All cells were grown in a humidified atmosphere of 95%
air/5% CO2 at 37 °C and experiments were performed on 4th and 5th passages generated from the frozen stock. Routine tests to exclude Mycoplasma in all cell lines were conducted several times
each year. Short tandem repeat analysis was conducted to ensure cells (MCF-7, A-431, PC3 and LNCaP) were of human origin in May 2013.
To generate tumours, 4- to 6-week-old male athymic nude mice (Jackson Laboratory, Bar Harbor, ME, USA) were injected subcutaneously in both flanks with 2 × 106 PC3 or LNCaP cells, and 4- to
6-week-old female athymic nude mice (Harlan Sprague–Dawley) with implanted 17β-estradiol tablets (60-day release; Innovative Research of America) were injected subcutaneously with 5 × 106
MCF-7 cells per side; non-estradiol-implanted female athymic nude mice were injected with 1 × 106 A-431 cells. There were six mice per treatment group (12 tumours) and each experiment was
repeated three times.
Docetaxel was obtained from Sanofi Inc (Laval, Quebec). Pantoprazole was purchased from the hospital pharmacy as a lyophilised powder and dissolved in 0.9% saline. EF5 was provided by the
National Cancer Institute as a powder and then dissolved in distilled water supplemented with 2.4% ethanol and 5% dextrose to make a 10-mM stock solution that was stored at room temperature.
Cy5-conjugated mouse anti-EF5 antibody was purchased from Dr Cameron Koch, University of Pennsylvania, Philadelphia, PA, USA. DiOC7 was purchased from AnaSpec (San Jose, CA, USA) and a
stock solution (2.5 mg ml−1) was made by dissolving in dimethyl sulphoxide; this stock was diluted 1 : 10 in phosphate-buffered saline and 10% Solutol HS 15 (Sigma-Aldrich, Oakville, ON,
Canada). γH2aX was recognised with a rabbit anti-human γH2aX primary antibody (Cell Signaling, Danvers, MA, USA). Cleaved caspase-3 was recognised with primary rabbit anti-human cleaved
caspase-3 antibody (Cell Signaling, Danvers, MA, USA). Ki67 was identified with primary rabbit anti-human Ki67 antibody (NovusBiologicals, Oakville, ON, Canada). LC3 was recognised with a
rabbit anti-human LC3 primary antibody and p62 was recognised with a rabbit anti-human p62 primary antibody (ABGENT, San Diego, CA, USA). Application of all primary antibodies was followed
by Cy3-conjugated goat anti-rabbit IgG secondary antibody and visualised using the Olympus fluorescent upright microscope.
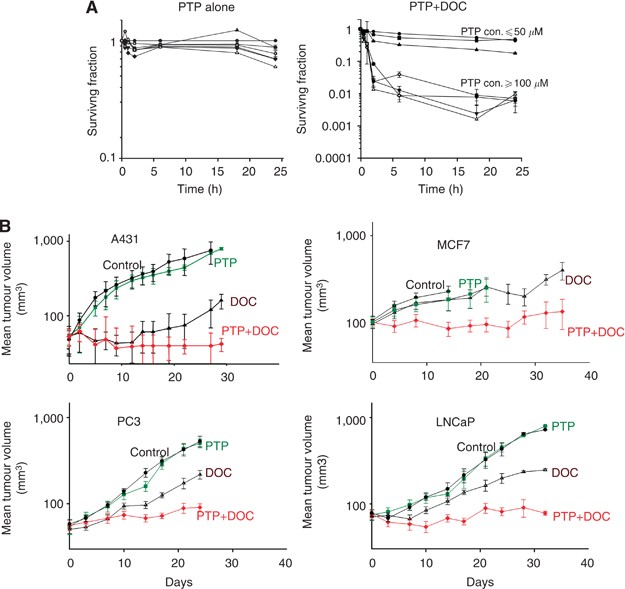
Single-cell suspensions were treated at 37 °C in 95% air+5% CO2 in stirred glass polyshell vials with or without 50 nM docetaxel in the presence or absence of pantoprazole at various
concentrations. Cells were counted and placed into a 13 ml tube at a concentration of 105cells ml−1. Serial dilutions were made to 104 and 103 cells ml−1 and each concentration was plated in
triplicate into six-well plates. Colonies generated 8–14 days later were stained with methylene blue and counted. The average colony count for each concentration was recorded and surviving
fraction was calculated using the following formula:
Mice bearing MCF-7, A-431, PC3 and LNCap tumours were divided into groups of six mice and treated weekly for 3 weeks with saline, docetaxel (15 mg kg−1 i.p.), pantoprazole (200 mg kg−1 i.p.)
or pantoprazole 2 h before docetaxel. These were maximum tolerated doses that caused minimal weight loss (Patel et al, 2013). All mice were ear-tagged and randomised to avoid bias. Two
perpendicular diameters of tumours growing in the flanks of mice were measured with a caliper every 2–3 days and treatment began once tumours reached a diameter of 5–8 mm. Measurements were
taken until tumours reached a maximum diameter of 1.5 cm or began to ulcerate, when mice were killed humanely. To minimise bias, we only continued measurements if at least 10 tumours were
available for assessment. Tumour volume was estimated using the formula: 0.5(ab2), where a is the longest diameter and b is the shortest diameter.
Mice bearing tumours of mean cross-sectional area 0.7–0.8 cm2 were treated with docetaxel (15 mg kg−1 i.p.), or pantoprazole (200 mg kg−1 i.p.) alone or 2 h before docetaxel. To detect
hypoxia and functional blood vessels, EF5 was injected i.p. ∼2 h before killing the mice (0.2 ml of a 10 mM stock per mouse) and the perfusion marker DiOC7 (1 mg kg−1) was injected i.v.1 min
before sacrifice. Mice were killed 10 min or 24 h after docetaxel injection and tumours were excised, embedded in OCT compound, frozen in liquid nitrogen and stored at −70 °C. Whole
cryostat sections (10 μm thick) were analysed and artifacts and regions of necrosis excluded. Tumour sections were first imaged for DiOC7 using a FITC filter set. Sections were then stained
for hypoxic regions using a Cy5-conjugated mouse anti-EF5 antibody (1 : 50) and with appropriate antibodies to one of the following biomarkers: γH2aX cleaved caspase-3, Ki67, LC3 and p62;
the sections were imaged using Cy3 filter set (530–560 nm excitation/573–746 nm emission). Image analysis and quantification of biomarker distribution in relation to blood vessels and
regions of hypoxia were performed as described previously (Fung et al, 2012).
Microtubule-associated protein 1 light chain 3 (LC3), a specific marker for autophagosome formation, has two forms, LC3-I and its proteolytic derivative LC3-II (molecular weight, 18 kDa and
16 kDa, respectively)(Xie and Klionsky, 2007). LC3-I is localised in the cytoplasm, whereas LC3-II binds to autophagosomes. Various stresses, such as hypoxia and starvation, stimulate the
conversion of LC3-I to LC3-II, and upregulation of LC3 expression(Mizushima et al, 2010). Relative autophagic flux can be measured by the levels of LC3-II degraded in autolysosomes in which
lysosomal hydrolases are functional (Klionsky et al, 2008). In addition to LC3, we also used p62/SQSTM1 as a marker of autophagy. The p62 protein serves as a link between LC3 and
ubiquitinated substrates but, unlike LC3, is degraded within the mature autolysosome (Bjorkoy et al, 2005). Thus, observation of increased p62 is indicative of a build-up of the protein due
to inhibition of lysosomal fusion to the autophagosome.
For western blot analysis, PC3 cells treated with or without pantoprazole in the presence or absence of bafilomycin A1 were lysed in RIPA buffer and centrifuged at 13 000 g at 4 °C for 30
min. Protein concentration in the supernatant was determined using a Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA) to quantify LC3-I and LC3-II and p62.
The PC3 cells were also transfected with a plasmid containing LC3-II tagged at the N terminus with green (GFP) and red fluorescent protein (RFP); this probe allows distinction of
autophagosomes (GFP+RFP+ yellow puncta) and autolysosomes (GFP-RFP+ red puncta), as GFP fluorescence is quenched in the acidic autolysosomes(Kimura et al, 2007). Cells showing red
fluorescence have increased autophagy whereas control cells show yellow fluorescence. Images were examined under a × 20 lens on an Olympus fluorescence microscope using standard filter sets
for GFP and RFP. The western blots and the number and spatial distribution of punctae were quantified using Image Pro software (Version premier 9).
The PC3 cells with knockdown of ATG7 and BECLIN1 (or both) were generated. Lentiviral shRNA (ATG7 and BECLIN1) constructs were purchased from Open Biosystems (RMM4534_019584 and
RMM4534_028835). The ATG7 and BECLIN1 shRNAs were transfected into PC3 cells, either alone or together with packaging plasmids following the manufacturer’s protocol (Invitrogen ViraPower
Lentiviral Expression Systems kit, Carlsbad, CA, USA). The silencing efficacy of the various shRNA was assessed by WB analysis of ATG7 and BECLIN1 proteins using polyclonal antibodies.
Animal experiments described in this paper were carried out using Animal Use Protocol (AUP1232.15, 09/05/14) approved by Princess Margaret Cancer Center, University Health Network (UHN)
Animal Care Committee under the guidelines of the Canadian Council on Animal Care.
One-way ANOVA, followed by Tukey’s post hoc test, determined statistical differences between treatment groups. P