Transcriptional regulation by p53: one protein, many possibilities
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The p53 tumor suppressor protein is a DNA sequence-specific transcriptional regulator that, in response to various forms of cellular stress, controls the expression of numerous
genes involved in cellular outcomes including among others, cell cycle arrest and cell death. Two key features of the p53 protein are required for its transcriptional activities: its ability
to recognize and bind specific DNA sequences and to recruit both general and specialized transcriptional co-regulators. In fact, multiple interactions with co-activators and co-repressors
as well as with the components of the general transcriptional machinery allow p53 to either promote or inhibit transcription of different target genes. This review focuses on some of the
salient features of the interactions of p53 with DNA and with factors that regulate transcription. We discuss as well the complexities of the functional domains of p53 with respect to these
interactions. SIMILAR CONTENT BEING VIEWED BY OTHERS TISSUE SPECIFICITY AND SPATIO-TEMPORAL DYNAMICS OF THE P53 TRANSCRIPTIONAL PROGRAM Article 08 February 2023 DETERMINANTS OF P53 DNA
BINDING, GENE REGULATION, AND CELL FATE DECISIONS Article Open access 29 June 2024 P53 TARGET ANKRA2 COOPERATES WITH RFX7 TO REGULATE TUMOR SUPPRESSOR GENES Article Open access 24 August
2024 INTRODUCTION P53 sits at the hub of an extremely complex network within the living cell. Multiple signals, perhaps the most well studied being DNA damage, lead to the functional
activation of p53 which can upregulate or downregulate numerous target genes, thus, initiating a series of events resulting in cell-cycle arrest, senescence or apoptosis among other outcomes
(reviewed in Vogelstein _et al_1, Prives and Hall2). The complexity of p53 as a protein mirrors the functional complexity of p53-dependent events. Active p53 protein exists in the cell as a
tetramer made of four identical subunits.3 Each monomer, in turn, consists of several well-defined domains including a multipartite N-terminal transactivation (TA) domain (residues 1–73) a
proline-rich region (63–97), the centrally located and highly conserved DNA-binding core domain (DBD) (residues 94–312) and within the C-terminus are located its tetramerization domain
(residues 324–355) followed by an unstructured basic domain (CTD) (residues 360–393). An additional layer to p53 complexity is added by the numerous post-translational modifications this
protein may undergo, the most important of which seem to be phosphorylation, acetylation and ubiquitination (reviewed in Brooks and Gu4, Appella and Anderson5). Site- and time-specific
modifications of certain p53 residues are likely to regulate its activity as well as its half-life although contradictory findings in the literature imply great complexity in this regard.
Though recent studies have demonstrated that p53 can also produce apoptosis through non-transcriptional processes that occur in the cytoplasm (reviewed in Erster and Moll6), in this review
we will focus on the roles and regulation of p53 as a transcriptional activator and repressor. From that vantage point p53 may be viewed as possessing two critical functions and their
related modules: DNA-binding and co-factor recruiting. We would then suggest that the other regions of p53 serve to assist these two primary modules in performing their functions. Since the
discovery of the first transcription targets of p53 in the early 1990s (_p21, GADD45, cyclin G, bax_ and others, reviewed in Ko and Prives7) the list of genes whose expression depends on p53
has grown dramatically and continues to expand. Such p53 targets have been identified either as single genes or using different screening techniques such as DNA microarrays or SAGE
technology. In the current review we will focus mainly on a number of aspects of the p53 protein's function as a transcription factor and its ability to modulate the transcription of
downstream genes in a positive and negative manner. We will also discuss some cis- and trans-elements participating in these processes. P53 is one of the most extensively scrutinized
mammalian regulatory genes to have been examined in recent decades. Given the confines of the format of this review, it has been therefore impossible to cite all of the excellent studies
that have focused on its roles as a transcription factor. We sincerely apologize in advance to those whose work we have unintentionally omitted in this article. P53 BINDING TO DNA THE P53
DNA CONSENSUS RECOGNITION ELEMENT (RE) To initiate the chain of events resulting in either inhibition of cell-cycle progression or programmed cell death, p53 must recognize and bind to its
recognition elements (REs) that are located in the vicinities of its target genes. In most cases, p53 REs are found within a few thousand base pairs of transcriptional start site and
frequently p53 target genes have at least two widely spaced p53 REs. The consensus p53 DNA RE consists of two pairs (half-sites) of head-to-head arranged pentamers,
5′-PuPuPuCA/TA/TGPyPyPy-3′ (Pu is purine, Py is pyrimidine) separated by 0–13 nucleotides.8 The sequence within the pentamers as well as the pentamers mutual orientation is the important
determinant that may influence both, p53 binding ability and activity.9, 10 Strong p53 REs have been shown to possess certain general features. The most important of them are (i) the
presence of highly conserved and obligate C and G residues (ii) either 0 or 1 bp spacing between the two half-sites, and (iii) no more than 3 non-consensus bases within RE.11, 12
Interestingly, the presence of AT residues in the middle of each half-site results in the strongest positive activation, whereas non-consensus bases are associated with low functionality
noticeably affecting p53-dependent transactivation.12 p53 may also recognize REs whose structure is somewhat different from the consensus RE. For instance, one that is weakly induced by p53
is AQP3, in which, the RE is made up of three pairs of pentamers.13 Another example is the p53 RE located within the promoter of the MDR1 gene (see below).14 A third is the microsatellite
region within the PIG3 gene that was shown to function even more effectively as a p53 RE than a more canonical binding site within the same promoter.15 Finally, Walter _et al._16 have
recently demonstrated the ability of p53 to recognize and bind to CTG·CAG trinucleotide repeats with the subsequent induction of topological alterations in DNA. It is noteworthy that among
p53-regulated genes their respective levels of activation or inhibition of transcription vary broadly. This is due, at least in part, to the variation within the individual p53 REs. When
tested in a yeast model system such variations resulted in up to 1000-fold difference in transactivation.10, 17 Yeast-based assays also have been used by recent studies to identify new
potential p53 REs and to evaluate the impact of genetic variation within the REs on p53-dependent transactivation.11 In addition to the discovery of a number of new p53 potential targets,
they established a link between particular p53 RE alleles and their influences on p53 transactivation. It should be noted, however, that such differences observed in yeast systems may not be
replicated in mammalian cells. P53 HAS TWO AUTONOMOUS DNA–BINDING DOMAINS THE P53 CORE DOMAIN The large central core DBD demonstrates high affinity toward the consensus RE (see above),
whereas the small, C-terminally located highly basic domain (CTD) has been shown to bind to DNA without sequence specificity (reviewed in Jayaraman and Prives18, Kim and Deppet19, Liu and
Kulesz-Martin20). On the basis of crystallographic data a model has been suggested in which each pentamer of the RE interacts with the DBDs comprising a p53 tetramer.21 The crystal structure
of the p53 core DBD revealed that its complex _β_-sheet sandwich and loop-sheet-helix motif are the sequence-specific DNA-binding determinants of the core. This model has been validated in
a number of NMR-based and biochemical studies.22, 23, 24 Among more than 18 000 currently known mutations in p53 that result in either partial or complete loss of its wild-type (wt)
functions, amino acids within the p53 DBD are by far the most frequent mutational targets.25 The mutations situated in the DBD can disrupt p53-specific DNA binding by several possible ways.
Mutations at some positions (e.g. R273) decrease the binding ability of p53 by simply eliminating direct contacts between the protein and DNA,21, 26 while some other mutations (e.g. R175,
G245, R249, R282) diminish binding by destabilizing the tertiary structure of p53 DBD.27, 28 Mutation at position 248 (R248), in addition to breaking DNA-protein contacts, also introduces
extensive structural changes into the DBD.27 Among these mutations are also those that lead to the loss or significant decrease in Zn2+ ions, normally chelated by C176 and H179 of the L2
loop and by C238 and C242 of the L3 loop.21 In fact, Zn2+ release from the wild-type DBD of p53 leads to a conformational change of L2 and L3 loops and facilitates conversion of an active
soluble protein into one that is insoluble and inactive.29 Importantly, the loss of Zn2+ ions is facilitated by elevating temperature, a finding which may provide an explanation of the
unusual loss-of-function phenomenon demonstrated by p53 at 37°C30 although the N-terminus has also been shown to be involved in stabilization of temperature-sensitive DNA binding by
wild-type31 and mutant32 forms of p53. Additionally, Butler and Loh33 have described the ability of some DBD mutants (G245S, R249S, R282Q) to facilitate the loss of p53 function by causing
DBD to cycle unusually rapidly between folded and unfolded states. During such cycling a fraction of DBD caught in a functionally inactive state continually increases. Recognition and making
contact with the different bases within a consensus RE requires a significant level of flexibility of the p53 DBD within a tetramer. The same flexibility, though, seems to be responsible
for the ability of some p53 mutants to bind and inactivate the other two p53 relatives, p63 and p73,34, 35, 36, 37 as well as to force wild-type p53 molecules (translated from the remaining
wild-type p53 allele) into a mutant conformation. Surprisingly, while the existence of even one mutated p53 allele in the cell results in a gain-of-function phenotype,38, 39 Chan _et al._40
found that at least three DNA-binding defective molecules of p53 are needed in order to effectively inactivate the tetramer. They reported that in their experiments the presence of one or
two mutated DBD molecules within a tetramer does not significantly reduce p53 transcriptional activation. Speculatively, the existence of partially active tetramers in the cell may lead to a
new phenotype via appearance of an unusual binding surface on a p53 molecule, which may change the pattern of coactivators/corepressors interacting with p53 or even promote new contacts.
Alternatively, mutations in the DBD may either result in the ability of p53 to bind selectively to target genes or (more speculatively) to some novel DNA sequences thus altering the normal
global transcriptional response to p53. THE P53 C-TERMINAL DNA-BINDING DOMAIN The small CTD of p53 initially attracted attention because of its ability to bind to nonspecific sequences in
DNA, and also its potential ability to directly regulate binding by the p53 sequence-specific DBD. Unstructured and rich in both serine and lysine residues, the CTD can be subjected to
several types of modification the most well studied being phosphorylation and acetylation.5, 41, 42. In addition, it has been suggested that C-terminal lysines of p53 may be involved in the
regulation of stability of p53, as they are subjected to MDM2-mediated ubiquitination. In fact, the same lysine residues are acetylated by histone acetyl transferases (HATs) which result in
p53 stabilization and activation.43, 44, 45 Finally, there are data that neddylation,44 methylation,46 and sumoylation (reviewed in Muller _et al._47) of the CTD can regulate p53 functions.
What is the meaning of such C-terminal modifications? It was originally observed that, using the electrophoretic mobility shift assay (EMSA), either binding by the C-terminal antibody PAb
421 or phosphorylation of the penultimate residue, S392 leads to increased ability of p53 to bind to DNA. It was hypothesized that after DNA damage, the CTD of p53 becomes modified by
phosphorylation leading to increased binding to its transcriptional target genes (Reviewed in Ahn and Prives48). This suggestion was extended to a proposal that upon such modification p53
normally in a latent state in cells undergoes a modification-induced allosteric conformational shift that somehow positively affects the ability of the core domain to bind to DNA. In fact,
_in vitro_ and _in vivo_ obtained data suggested that not only phosphorylation but CTD acetylation also enhances the sequence-specific DNA binding of p53.49, 50, 51 This concept was also
supported by the observation that deletion of the C-terminus increases its ability to bind to DNA using similar experimental conditions. The allosteric model was subsequently challenged by
Anderson _et al._ who showed that the interactions of p53 with PAb 421 or long DNA molecules had respectively stimulatory or inhibitory effects on its ability to bind to a short
RE-containing oligonucleotide.52 They proposed that the C-terminus binding to long DNA interferes with the ability of the core to bind to DNA and that modification of the C-terminus prevents
such interference by blocking its ability to bind to nonspecific DNA. Their hypothesis was supported by NMR analysis that showed that both full length and C-terminally deleted versions of
p53 have essentially similar structures53 and by experiments from the group of Fersht showing that when fully acetylated the C-terminus completely loses DNA binding capacity.54 Relevantly,
Espinosa and Emerson reported that although acetylation appears to increase the p53-binding affinity toward short double-stranded DNA fragments it does not affect ability of p53 to bind to
its specific RE in the context of chromatin or longer DNA molecules.55 It has become clear that evaluating the p53 DNA binding using short double-strand oligonucleotides does not provide a
complete picture. Importantly as well, quantitative chromatin immunoprecipitation analysis has failed to demonstrate allosteric regulation of DNA binding of p53: neither of the two groups of
p53 target genes – with relatively high promoter occupancy _in vivo_ or with much weaker DNA binding exhibit significant enrichment in p53 binding after genotoxic stress.56 Finally, it was
recently shown that substitution of all p53 CTD lysines for arginines does not significantly affect the activities or stability of p53 in a mouse model.57, 58 Though controversial, the
importance of the CTD in p53 functioning has been continuing to reveal itself in a number of more recent publications. McKinney and Prives,59 Fojta _et al._,60 and Palecek _et al._61 have
shown that the CTD is important for binding to various non-linear DNAs. In another study, McKinney _et al._62 have demonstrated the involvement of CTD in the ability of p53 to diffuse
linearly on DNA. Both, McKinney _et al._62 and Liu _et al._63 showed as well that the CTD is required for efficient promoter activation _in vivo_ by p53. Moreover, Harms and Chen64 have
recently reported that the CTD inhibits p53-dependent induction of IGFBP3 gene. In this case it is HAT-related inhibitory activity, but not p53 DNA binding ability, that is bound to CTD. The
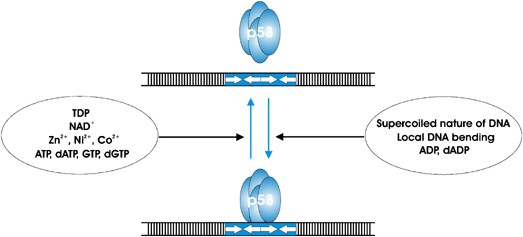
ability of p53 to interact with DNA sequences can be modulated by the presence of small molecules. First, as has been demonstrated, elevating concentrations of transition metals such as
Zn2+, Ni2+, and Co2+ inhibit p53 binding to the DNA RE.65 Of these three, Zn2+ ions have had the most devastating effect when added in the concentration 5–20 _μ_M. Second, the presence of
ADP or dADP has been found to stabilize p53-DNA complexes, while addition of ATP dATP, GTP or dGTP apparently can facilitate the release of p53 from such complexes.66 Two other molecules,
thiamine diphosphate (TDP) and nicotinamide adenine dinucleotide (NAD+) have been recently shown to bind to p53, induce conformational changes, and inhibit DNA binding.67 The authors propose
that the level and rate of NAD+ synthesis may directly modify p53 DNA specificity. In addition to small molecules, other factors, like the supercoiled nature of the DNA or local DNA
bending, may facilitate p53-DNA interactions.59, 61 Importantly, it has been demonstrated earlier that there is a direct correlation between the stability of p53-DNA complexes and the
ability of the DNA to be bent by the p53 DBD upon binding.68 A summary of the above regulatory effectors is shown in Figure 1. For the purpose of simplicity and clarity we have omitted from
Figure 1 the numerous post-translational modifications and protein factors previously reported to regulate p53 DNA binding. Furthermore, as discussed above some of these modifications (e.g.
acetylation) and proteins have been shown to either stimulate or inhibit p53 DNA binding depending on the experimental context. THE COMPLEXITY OF THE P53 TRANSACTIVATION DOMAIN The first
suggestion that p53 functions as a transcriptional regulator came from that observation that its N-terminus (TA) when fused to a heterologous DNA-binding domain can serve as an activation
domain in yeast. Owing to the relatively high number of paired aspartate and glutamate residues located within it, this domain belongs to the class of ‘acidic’ activation domains that are
known to interact with a number of proteins that populate the general transcription machinery (reviewed in Ko and Prives7). Although the conclusions based on these observations that p53 is a
transcriptional activator turned out to be correct, formal proof that p53 is a sequence-specific transcriptional activator was only obtained when full-length p53 was studied in that
context. The TA domain is one of the several unstructured regions within p53.69 Though the entire N-terminal region of p53 is natively unfolded, and that seems to be its functional state,70
NMR-based studies have found several small ‘islands’ of secondary structure within the TA: the helix formed by residues Thr18–Leu26, and the two turns are shaped by residues Met40–Met44 and
Asp48–Trp53, respectively.71 It has been suggested that these regions may represent recognition scaffolds for p53-interacting proteins. Interestingly, as is the case with other activation
domains, single-point mutations within the TA domain do not demonstrate such a devastating effect as in the case of DBD.72 Thus, the overall shape of the TA polypeptide chain may not be
affected by single amino-acid changes and would seem to be more important for recognition and protein interaction rather than any specific residues. Nevertheless, simultaneous mutation of
two hydrophobic residues Leu22 and Trp23 markedly impairs (but does not abolish) transactivation by p53.72 Interestingly, mutation of residues Trp53 and Phe54 appears to affect specifically
the ability of p53 to regulate some of its pro-apoptotic target genes in some settings.73 Though the results obtained by mutational analysis of p53 TA have demonstrated the presence of two
highly important subdomains within it and set its boundaries between the residues 1–42 and 43–73,74 several other regions of TA (as well as amino acids located within them) have been shown
to participate in the process of transcriptional regulation. For example, some co-activators have been shown to interact with p53 within residues 63–97.75 Residues 63–97 span a proline-rich
region shown to be important for the Pin-induced modification of the TA structure,76, 77 and to interact directly with p300.78 One more functionally divergent region within the N-terminal
part of p53 is a recently identified repression domain.79 As defined by mutation analysis, it is located between proline-rich domain and DNA-core domain (residues 100–116). Importantly, this
domain can function as an independent heterologous repressor, and has been shown to decrease VP16-driven activation up to 20 times in human embryonal carcinoma cells, when being fused to
VP16 transactivator.79 As wild-type p53 is found to be overexpressed in some type of tumors, it has been proposed that the newly identified domain may play an important role in repression of
the basal activity of p53. Factors that are or can be specifically recruited by this domain, and that might be essential for the repression phenomenon found, are not known at present. Thus,
these and other experiments further highlight the likelihood that transcriptional regulation by p53 involves several different regions within its N-terminus. In addition to the multipartite
nature of the activation domain of p53, there are three other reasons why this is an exceeding complex region. First, it is the region to which its negative regulator Mdm2 binds and targets
p53 for both repression of its transcriptional regulation functions and also for proteasome-mediated degradation (reviewed in Bond _et al._80). Second, it was reported to possess a
previously unknown nuclear export signal spanning the residues 11–27.81 Finally, the N-terminus of p53 is extensively modified. Site-specific phosphorylation of the TA domain is important
for both, p53 stability82, 83 and activity50, 84, 85, 86, 87 and has been shown to be interdependent.88 There are also qualitative differences in phosphorylation in response to different
genotoxic agents. For example, phosphorylation of Thr18 has been shown to be stronger when cells are exposed to ionizing radiation and adriamycin rather than to UV light while
phosphorylation at Ser33, Ser37, and Ser46 has been more pronounced in case of UV treatment.88 As in the case with p53 CTD modifications, the role of site-specific phosphorylation of the key
serines within TA domain has been challenged by earlier89 and some more recent data. Thompson _et al._90 using Nutlin-3, a recently developed small molecule MDM2 antagonist, have not
detected any induction in the phosphorylation of p53 and yet both its DNA sequence-specific binding and ability to transactivate its target genes were unaltered when compared to p53 that is
induced after forms of DNA damage that lead to extensive N-terminal modifications. TRANSCRIPTION REGULATION BY P53 Several views exist on the mechanism by which p53 may promote
transcription. The first one is built on the assumption that the promoter region of the gene to be activated by p53 is usually not accessible to the general transcription factors and RNA
polymerase. In this scenario binding of p53 to its REs within a promoter would facilitate promoter opening via recruiting either chromatin remodeling factors (CRF)91, 92 (though see
reference Hill _et al._93) or histone transacetylases (HAT)94, 95, 96, 97 and/or methyltransferases98 (Figure 2). This view has been validated recently in a significant number of studies.
Physical and functional interactions between p53 and p300 HAT have been well documented.49, 55, 94, 95, 96, 97 The involvement of PRMT1 and CARM1 methyltransferases in p53 function has been
also demonstrated in the _in vitro_ study utilizing a chromatin template with GADD45 p53 RE.98 Thus, histone modifications and the subsequent alterations in chromatin structure and function
seem to be one of the major outcomes of p53 binding to the RE. In addition, p53 has been shown to facilitate formation of preinitiation complex via direct interactions with the components of
Mediator complex.99, 100 p53 can also stimulate transcription by enhancing the recruitment of the basal transcription factors, such as TFIIA and TFIID through the direct interactions with
them, and by inducing conformational change(s) in these complexes.7, 101 Another view of p53-depending transcriptional activation is based on the observation that the regions of chromatin
within the vicinity of several p53 responsive promotes (including GADD45 and MDM2) exist in open conformations regardless of the conditions (normal _versus_ genotoxic).102, 103, 104
Constitutive hypersensitivity of these promoters to DNase I led to the suggestion that they might be nucleosome-free, and do not require significant chromatin alterations to become
activated. By analogy with some genes, which are regulated by promoter proximal pausing and factors facilitating reinitiation of stalled RNA Polymerase, p53 has been suggested to induce
reinitiation of transcription.104 For further evaluation this model clearly requires more data. The subject of p53-dependent transcription activation, including an up-to-date list of
positively regulated genes, has been more than adequately covered in a series of recent reviews.105, 106 NEGATIVE REGULATION OF TRANSCRIPTION BY P53 Although well established as a
transcriptional activator, p53 has also been shown to suppress the transcription of certain genes. Indeed, in addition to identification of multiple targets that are activated by p53,
expression arrays have indicated that significant numbers of genes are downregulated after induction of p53 in many cases.107, 108, 109, 110 The molecular background of negative regulation
of gene expression by p53 seems to be more diverse. Interestingly, earlier reports have suggested that transcriptional repression by p53 is the key activity that is required for its ability
to induce cell death.7 p53 can efficiently inhibit transcription driven by all three mammalian RNA polymerases (Pol I, Pol II and Pol III). With respect to Pol II inhibition, several
mechanisms have been documented (Figure 3). These include repression of transcription activators by physical interaction with and preventing them from activating the promoter111, 112, 113,
114, 115, 116 or by displacing them from the adjusting or overlapping binding sites within the promoter,117, 118, 119, 120 interference with the assembly of transcription machinery,7, 121
repression through the recruitment of histone deacetylase (HDAC) and, possibly, other chromatin modifying factors,122, 123, 124 and finally through novel REs with the unique architecture
that dictates the outcome of p53 binding.14, 125 Combination of two of the above mechanisms has been also documented.126 It should be noted, that the p53-dependent transcriptional repression
that depends on HDAC recruitment, has perhaps the most complicated scenario, and in many cases it is mediated by the presence of additional protein(s). The known mediators include
mSin3a,122, 127 SnoN128 and p52.129 In certain cases p53-induced repression seems to depend on the activation of p53-dependent targets. p53 mediated repression of Chk1,130 or Cdc2, Cyclin
A2, survivin and some other genes may occur indirectly through the transcriptional activation of p21.131 In such cases p21 alone is sufficient to inhibit the transcription to the same extent
as p53. Whatever the nature of p53-dependent repression, it is clear that it requires functionally active p53 protein. Surprisingly, mutations in almost all domains of p53 – TA, DBD,
proline-rich and CTD – may abrogate its inhibitory ability, suggesting the specific role for each of the domains during inhibition. For example, p53 TA-deficient mutants lose the ability to
repress the transcription of the Map4 gene,132 whereas a p53 mutant lacking the proline-rich domain cannot repress a series of promoters as efficiently as wild-type p53.133 This domain is
also important for p53-Sin3a-mediated transcription inhibition.75, 122 The C-terminal domain of p53 is required for the interaction between p53 and Ets-1 protein, which is necessary for TXSA
repression.112 It is intriguing that the presence of a functionally active DBD is required for the inhibition to occur even if binding of p53 to the RE is not necessary. In this context it
should be noted that promoters of some downregulated genes have one or more potential p53 REs, not participating in p53-mediated regulation.112, 134, 135 The human small nuclear RNA U1 gene
has been shown to contain a high-affinity RE within its promoter which nevertheless seems to be dispensable for p53-mediated repression of U1.135 Whether recognition of such ‘latent’ REs
depends on the intracellular conditions and/or external stimuli, remains to be found. Recently, transcriptional repression that results from the binding of p53 to a novel type of ‘repression
site’ RE has been described in the MDR1 promoter.14, 125 Interestingly, it is orientation of pentamers – ‘head-to-tail’, instead of ‘head-to-head’ – within the RE that dictates the type of
p53 activity on this promoter. The change of the orientation back to ‘normal’ results in the significant activation of MDR1. The same authors propose that the inhibition of several other
genes, namely, cyclin A, cyclin B1, and ARF, may be driven by the same mechanism, as identical head-to-tail REs have been identified within their promoters.14 For cyclin B1, this might be an
alternative mechanism, as its repression has been shown to depend on p21/CDKN1.131 Surprisingly, two p53 homologues, p63 and p73, do not recognize the Mdr1 repression site RE and are unable
to inhibit transcription at MDR1 promoter, suggesting some unique properties for DBD domain of each type.125 Taking into account that activity of p53-repressed genes usually correlates with
cell proliferation or malignant progression, it is not surprising that p53 may also suppress such activities in a more general way by targeting the components of protein biosynthesis
machinery. p53 does it by inhibiting the expression of RNA Pol I136, 137 and RNA Pol III135, 138, 139 transcribed genes. Pol I-driven transcription is repressed by p53 interference with the
assembly of a productive initiation complex on the rRNA promoter,136, 137 whereas transcription of tRNAs seems to be downregulated directly, through the interaction with the components of
Pol III transcription machinery,138, 139 and indirectly, by p53-dependent degradation of TFIIIB.140 Both, rRNA and tRNA synthesis is significantly elevated in fibroblasts from p53-knockout
mice.137, 141 CONCLUDING REMARKS The ability of p53 to regulate transcription of a number of genes in response to different genotoxic signals lies at the center of its function as a major
tumor suppressor. The ongoing process of identification of numerous p53-regulated genes has been slowly revealing the multifaceted and somewhat knotted mechanism by which p53 exerts its
functions in cells. p53 clearly demonstrates the interdependence of the many ‘discrete’ steps in the process of transcription regulation, and their importance for the final outcome. Yet,
despite the myriad of studies delving into the mechanisms by which p53 regulates its targets there is still much more to learn about this fascinating protein. Looking ahead into the future
we envisage many new avenues to be explored, some of which will require novel or more refined technologies. The contacts made by p53 with components of the transcriptional machinery need to
be determined at the atomic level. We would also hope to gain a dynamic or kinetic view of p53 as it regulates transcriptional target genes. How will p53 be affected by the other p53
isoforms recently identified142, 143 or by the presence of isoforms encoded by its sibling genes, p63 and p73 (reviewed in Harms and Chen144)? Finally, the ‘Holy Grail’ of p53 research still
lies ahead: capitalizing upon past and future basic research discoveries to improve diagnosis and treatment of cancer patients. ABBREVIATIONS * AQP3: aquaporin 3 * ARF: alternate open
reading frame * (d)ADP: (deoxy)adenosine diphosphate * (d)ATP: (deoxy)adenosine triphosphate * CARM1: coactivator-associated arginine methyltransferase 1 * Cdc2: cell division cycle 2 *
Chk1: checkpoint kinase 1 * CRF: chromatin remodeling factor * CTD: C-terminal basic domain * DBD: DNA-binding core domain * EMSA: electrophoretic mobility shift assay * Ets-1: V-ets
erythroblastosis virus E26 oncogene homolog 1 (avian) * GADD45: growth arrest and DNA damage inducible gene 45 * (d)GTP: (deoxy)guanosine triphosphate * HAT: histone transacetylase * HDAC:
histone deacetylase * IGFBP3: insulin-like growth factor binding protein 3 * MDM2: mouse double minute 2 * MDR1: P-glycoprotein (multidrug resistance 1) * NAD+: nicotinamide adenine
dinucleotide * NMR: nuclear magnetic resonance * PIG3: p53-induced gene 3 * Pin: peptidyl-prolyl cis/trans isomerase * PRMT1: protein arginine methyltransferase 1 * RE: p53 DNA consensus
recognition element * RNA Pol I/II/III: DNA-dependent RNA Polymerase I or II or III * rRNA: ribosomal RNA * tRNA: transfer RNA * SAGE: serial analysis of gene expression * SnoN: SKI-like *
mSin3a: transcriptional regulator SIN3A * TA: N-terminal transactivation domain * TDP: thiamine diphosphate * TFIIA: transcription factor IIA * TFIID: transcription factor IID * TFIIIB:
transcription factor IIIB * TXSA: thromboxane synthase * VP16: Herpes simplex virus transactivator VP16 * p21/CDKN1: cyclin-dependent kinase inhibitor 1A * p52, nuclear factor of kappa light
polypeptide gene enhancer in B-cells 2: p49/p100 * p53: tumor suppressor p53 * p300: transcriptional co-activator protein p300 REFERENCES * Vogelstein B, Lane D and Levine AJ (2000) Surfing
the p53 network. _Nature_ 408: 307–310. CAS PubMed Google Scholar * Prives C and Hall PA (1999) The p53 pathway. _J. Pathol._ 187: 112–126. Article CAS PubMed Google Scholar *
Jeffrey PD, Gorina S and Pavletich NP (1995) Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. _Science_ 267: 1498–1502. Article CAS PubMed
Google Scholar * Brooks CL and Gu W (2003) Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. _Curr. Opin. Cell Biol._ 15: 164–171. Article CAS
PubMed Google Scholar * Appella E and Anderson CW (2001) Post-translational modifications and activation of p53 by genotoxic stresses. _Eur. J. Biochem._ 268: 2764–2772. Article CAS
PubMed Google Scholar * Erster S and Moll UM (2005) Stress-induced p53 runs a transcription-independent death program. _Biochem. Biophys. Res. Commun._ 331: 843–850. Article CAS PubMed
Google Scholar * Ko LJ and Prives C (1996) p53: puzzle and paradigm. _Genes Dev._ 10: 1054–1072. Article CAS PubMed Google Scholar * Hoh J, Jin S, Parrado T, Edington J, Levine AJ and
Ott J (2002) The p53MH algorithm and its application in detecting p53-responsive genes. _Proc. Natl. Acad. Sci. USA_ 99: 8467–8472. Article CAS PubMed PubMed Central Google Scholar *
Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH, Tom E, Mack DH and Levine AJ (2000) Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. _Genes Dev._
14: 981–993. Article CAS PubMed PubMed Central Google Scholar * Inga A, Storici F, Darden TA and Resnick MA (2002) Differential transactivation by the p53 transcription factor is highly
dependent on p53 level and promoter target sequence. _Mol. Cell. Biol._ 22: 8612–8625. Article CAS PubMed PubMed Central Google Scholar * Tomso DJ, Inga A, Menendez D, Pittman GS,
Campbell MR, Storici F, Bell DA and Resnick MA (2005) Functionally distinct polymorphic sequences in the human genome that are targets for p53 transactivation. _Proc. Natl. Acad. Sci. USA_
102: 6431–6436. Article CAS PubMed PubMed Central Google Scholar * Resnick MA, Tomso D, Inga A, Menendez D and Bell D (2005) Functional diversity in the gene network controlled by the
master regulator p53 in humans. _Cell Cycle_ 4: 1026–1029. Article CAS PubMed Google Scholar * Zheng X and Chen X (2001) Aquaporin 3, a glycerol and water transporter, is regulated by
p73 of the p53 family. _FEBS Lett._ 489: 4–7. Article CAS PubMed Google Scholar * Johnson RA, Ince TA and Scotto KW (2001) Transcriptional repression by p53 through direct binding to a
novel DNA element. _J. Biol. Chem._ 276: 27716–27720. Article CAS PubMed Google Scholar * Contente A, Dittmer A, Koch MC, Roth J and Dobbelstein M (2002) A polymorphic microsatellite
that mediates induction of PIG3 by p53. _Nature Genet._ 30: 315–320. Article PubMed Google Scholar * Walter K, Warnecke G, Bowater R, Deppert W and Kim EL (2005) tumor suppressor p53
binds with high affinity to CTG. CAG trinucleotide repeats and induces topological alterations in mismatched duplexes. _J. Biol. Chem._ 280: 42497–42507. Article CAS PubMed Google Scholar
* Resnick MA and Inga A (2003) Functional mutants of the sequence-specific transcription factor p53 and implications for master genes of diversity. _Proc. Natl. Acad. Sci. USA_ 100:
9934–9939. Article CAS PubMed PubMed Central Google Scholar * Jayaraman L and Prives C (1999) Covalent and noncovalent modifiers of the p53 protein. _Cell Mol. Life Sci._ 55: 76–87.
Article CAS PubMed Google Scholar * Kim E and Deppert W (2006) The versatile interactions of p53 with DNA: when flexibility serves specificity. _Cell Death Differ_. 13: 885–889. Article
CAS PubMed Google Scholar * Liu Y and Kulesz-Martin MF (2006) Sliding into home: facilitated p53 search for targets by the basic DNA binding domain. _Cell Death Differ_. 13: 881–884.
Article CAS PubMed Google Scholar * Cho Y, Gorina S, Jeffrey PD and Pavletich NP (1994) Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations.
_Science_ 265: 346–355. Article CAS PubMed Google Scholar * Klein C, Planker E, Diercks T, Kessler H, Kunkele KP, Lang K, Hansen S and Schwaiger M (2001) NMR spectroscopy reveals the
solution dimerization interface of p53 core domains bound to their consensus DNA. _J. Biol. Chem._ 276: 49020–49027. Article CAS PubMed Google Scholar * Rippin TM, Freund SM, Veprintsev
DB and Fersht AR (2002) Recognition of DNA by p53 core domain and location of intermolecular contacts of cooperative binding. _J. Mol. Biol._ 319: 351–358. Article CAS PubMed Google
Scholar * McLure KG and Lee PW (1998) How p53 binds DNA as a tetramer. _EMBO J._ 17: 3342–3350. Article CAS PubMed PubMed Central Google Scholar * Data available at International
Agency for Research on Cancer TP53, database R8, available at www.iarc.fr/p53. * Bullock AN, Henckel J, DeDecker BS, Johnson CM, Nikolova PV, Proctor MR, Lane DP and Fersht AR (1997)
Thermodynamic stability of wild-type and mutant p53 core domain. _Proc. Natl. Acad. Sci. USA_ 94: 14338–14342. Article CAS PubMed PubMed Central Google Scholar * Wong KB, DeDecker BS,
Freund SM, Proctor MR, Bycroft M and Fersht AR (1999) Hot-spot mutants of p53 core domain evince characteristic local structural changes. _Proc. Natl. Acad. Sci. USA_ 96: 8438–8442. Article
CAS PubMed PubMed Central Google Scholar * Bullock AN, Henckel J and Fersht AR (2000) Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of
mutant states for rescue in cancer therapy. _Oncogene_ 19: 1245–1256. Article CAS PubMed Google Scholar * Butler JS and Loh SN (2003) Structure, function, and aggregation of the
zinc-free form of the p53 DNA binding domain. _Biochemistry_ 42: 2396–2403. Article CAS PubMed Google Scholar * Foster BA, Coffey HA, Morin MJ and Rastinejad F (1999) Pharmacological
rescue of mutant p53 conformation and function. _Science_ 286: 2507–2510. Article CAS PubMed Google Scholar * Hansen S, Hupp TR and Lane DP (1996) Allosteric regulation of the
thermostability and DNA binding activity of human p53 by specific interacting proteins. CRC Cell Transformation Group. _J. Biol. Chem._ 271: 3917–3924. Article CAS PubMed Google Scholar
* Friedlander P, Legros Y, Soussi T and Prives C (1996) Regulation of mutant p53 temperature-sensitive DNA binding. _J. Biol. Chem_ 271: 25468–25478. Article CAS PubMed Google Scholar *
Butler JS and Loh SN (2005) Kinetic partitioning during folding of the p53 DNA binding domain. _J. Mol. Biol._ 350: 906–918. Article CAS PubMed Google Scholar * Di Como CJ, Gaiddon C and
Prives C (1999) p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. _Mol. Cell. Biol._ 9: 1438–1449. Article Google Scholar * Gaiddon C, Lokshin M, Ahn J, Zhang T
and Prives C (2001) A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. _Mol. Cell. Biol._ 21: 1874–1887. Article
CAS PubMed PubMed Central Google Scholar * Bensaad K, Le Bras M, Unsal K, Strano S, Blandino G, Tominaga O, Rouillard D and Soussi T (2003) Change of conformation of the DNA-binding
domain of p53 is the only key element for binding of and interference with p73. _J. Biol. Chem._ 278: 10546–10555. Article CAS PubMed Google Scholar * Marin MC, Jost CA, Brooks LA, Irwin
MS, O'Nions J, Tidy JA, James N, McGregor JM, Harwood CA, Yulug IG, Vousden KH, Allday MJ, Gusterson B, Ikawa S, Hinds PW, Crook T and Kaelin Jr WG (2000) A common polymorphism acts as
an intragenic modifier of mutant p53 behaviour. _Nat. Genet._ 25: 47–54. Article CAS PubMed Google Scholar * Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA,
Terzian T, Caldwell LC, Strong LC, El-Naggar AK and Lozano G (2004) Gain of function of a p53 hot spot mutation in a mouse model of Li–Fraumeni syndrome. _Cell_ 119: 861–872. Article CAS
PubMed Google Scholar * Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D and Jacks T (2004) Mutant p53 gain of function in two mouse models of Li–Fraumeni syndrome.
_Cell_ 119: 847–860. Article CAS PubMed Google Scholar * Chan WM, Siu WY, Lau A and Poon RY (2004) How many mutant p53 molecules are needed to inactivate a tetramer? _Mol. Cell. Biol._
24: 3536–3551. Article CAS PubMed PubMed Central Google Scholar * Ljungman M, O'Hagan HM and Paulsen MT (2001) Induction of ser15 and lys382 modifications of p53 by blockage of
transcription elongation. _Oncogene_ 20: 5964–5971. Article CAS PubMed Google Scholar * Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E and Yao TP (2001) p300/CBP-mediated p53
acetylation is commonly induced by p53-activating agents and inhibited by MDM2. _EMBO J._ 20: 1331–1340. Article CAS PubMed PubMed Central Google Scholar * Rodriguez MS, Desterro JM,
Lain S, Lane DP and Hay RT (2000) Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. _Mol. Cell. Biol._ 20: 8458–8467. Article CAS PubMed PubMed
Central Google Scholar * Xirodimas DP, Saville MK, Bourdon JC, Hay RT and Lane DP (2004) Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. _Cell_ 118: 83–97.
Article CAS PubMed Google Scholar * Li M, Luo J, Brooks CL and Gu W (2002) Acetylation of p53 inhibits its ubiquitination by Mdm2. _J. Biol. Chem._ 277: 50607–50611. Article CAS PubMed
Google Scholar * Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, Barlev NA and Reinberg D (2004) Regulation of p53 activity
through lysine methylation. _Nature_ 432: 353–360. Article CAS PubMed Google Scholar * Muller S, Ledl A and Schmidt D (2004) SUMO: a regulator of gene expression and genome integrity.
_Oncogene_ 23: 1998–2008. Article CAS PubMed Google Scholar * Ahn J and Prives C (2001) The C-terminus of p53: the more you learn the less you know. _Nat. Struct. Biol._ 8: 730–732.
Article CAS PubMed Google Scholar * Gu W and Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. _Cell_ 90: 595–606. CAS PubMed
Google Scholar * Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW and Appella E (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade.
_Genes Dev._ 12: 2831–2841. CAS PubMed PubMed Central Google Scholar * Luo J, Li M, Tang Y, Laszkowska M, Roeder RG and Gu W (2004) Acetylation of p53 augments its site-specific DNA
binding both _in vitro_ and _in vivo_. _Proc. Natl. Acad. Sci. USA_ 101: 2259–2264. Article CAS PubMed PubMed Central Google Scholar * Anderson ME, Woelker B, Reed M, Wang P and
Tegtmeyer P (1997) Reciprocal interference between the sequence-specific core and nonspecific C-terminal DNA binding domains of p53: implications for regulation. _Mol. Cell. Biol._ 17:
6255–6264. Article CAS PubMed PubMed Central Google Scholar * Ayed A, Mulder FA, Yi GS, Lu Y, Kay LE and Arrowsmith CH (2001) Latent and active p53 are identical in conformation. _Nat.
Struct. Biol._ 8: 756–760. Article CAS PubMed Google Scholar * Friedler A, Veprintsev DB, Freund SM, von Glos KI and Fersht AR (2005) Modulation of binding of DNA to the C-terminal
domain of p53 by acetylation. _Structure (Cambridge)_ 13: 629–636. Article CAS Google Scholar * Espinosa JM and Emerson BM (2001) Transcriptional regulation by p53 through intrinsic
DNA/chromatin binding and site-directed cofactor recruitment. _Mol. Cell_ 8: 57–69. Article CAS PubMed Google Scholar * Kaeser MD and Iggo RD (2002) Chromatin immunoprecipitation
analysis fails to support the latency model for regulation of p53 DNA binding activity _in vivo_. _Proc. Natl. Acad. Sci. USA_ 99: 95–100. Article CAS PubMed Google Scholar * Krummel KA,
Lee CJ, Toledo F and Wahl GM (2005) The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. _Proc. Natl. Acad.
Sci. USA_ 102: 10188–10193. Article CAS PubMed PubMed Central Google Scholar * Feng L, Lin T, Uranishi H, Gu W and Xu Y (2005) Functional analysis of the roles of posttranslational
modifications at the p53 C terminus in regulating p53 stability and activity. _Mol. Cell Biol._ 25: 5389–5395. Article CAS PubMed PubMed Central Google Scholar * McKinney K and Prives C
(2002) Efficient specific DNA binding by p53 requires both its central and C-terminal domains as revealed by studies with high-mobility group 1 protein. _Mol. Cell. Biol._ 22: 6797–6808.
Article CAS PubMed PubMed Central Google Scholar * Fojta M, Pivonkova H, Brazdova M, Nemcova K, Palecek J and Vojtesek B (2004) Investigations of the supercoil-selective DNA binding of
wild type p53 suggest a novel mechanism for controlling p53 function. _Eur. J. Biochem._ 271: 3865–3876. Article CAS PubMed Google Scholar * Palecek E, Brazda V, Jagelska E, Pecinka P,
Karlovska L and Brazdova M (2004) Enhancement of p53 sequence-specific binding by DNA supercoiling. _Oncogene_ 23: 2119–2127. Article CAS PubMed Google Scholar * McKinney K, Mattia M,
Gottifredi V and Prives C (2004) p53 linear diffusion along DNA requires its C terminus. _Mol. Cell_ 16: 413–424. Article CAS PubMed Google Scholar * Liu Y, Lagowski JP, Vanderbeek GE
and Kulesz-Martin MF (2004) Facilitated search for specific genomic targets by p53 C-terminal basic DNA binding domain. _Cancer Biol. Ther._ 3: 1102–1108. Article CAS PubMed Google
Scholar * Harms KL and Chen X (2005) The C terminus of p53 family proteins is a cell fate determinant. _Mol. Cell. Biol._ 25: 2014–2030. Article CAS PubMed PubMed Central Google Scholar
* Palecek E, Brazdova M, Cernocka H, Vlk D, Brazda V and Vojtesek B (1999) Effect of transition metals on binding of p53 protein to supercoiled DNA and to consensus sequence in DNA
fragments. _Oncogene_ 18: 3617–3625. Article CAS PubMed Google Scholar * Okorokov AL and Milner J (1999) An ATP/ADP-dependent molecular switch regulates the stability of p53-DNA
complexes. _Mol. Cell. Biol._ 19: 7501–7510. Article CAS PubMed PubMed Central Google Scholar * McLure KG, Takagi M and Kastan MB (2004) NAD+ modulates p53 DNA binding specificity and
function. _Mol. Cell. Biol._ 24: 9958–9967. Article CAS PubMed PubMed Central Google Scholar * Nagaich AK, Appella E and Harrington RE (1997) DNA bending is essential for the
site-specific recognition of DNA response elements by the DNA binding domain of the tumor suppressor protein p53. _J. Biol. Chem._ 272: 14842–14849. Article CAS PubMed Google Scholar *
Bell S, Klein C, Muller L, Hansen S and Buchner J (2002) p53 contains large unstructured regions in its native state. _J. Mol. Biol._ 322: 917–927. Article CAS PubMed Google Scholar *
Dawson R, Muller L, Dehner A, Klein C, Kessler H and Buchner J (2003) The N-terminal domain of p53 is natively unfolded. _J. Mol. Biol._ 332: 1131–1141. Article CAS PubMed Google Scholar
* Lee H, Mok KH, Muhandiram R, Park KH, Suk JE, Kim DH, Chang J, Sung YC, Choi KY and Han KH (2000) Local structural elements in the mostly unstructured transcriptional activation domain
of human p53. _J. Biol. Chem._ 275: 29426–29432. Article CAS PubMed Google Scholar * Lin JY, Chen JD, Elenbaas B and Levine AJ (1994) Several hydrophobic amino acids in the p53
amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. _Gene Dev._ 8: 1235–1246. Article CAS PubMed Google Scholar *
Candau R, Scolnick DM, Darpino P, Ying CY, Halazonetis TD and Berger SL (1997) Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for
activity. _Oncogene_ 15: 807–816. Article CAS PubMed Google Scholar * Chang J, Kim DH, Lee SW, Choi KY and Sung YC (1995) Transactivation ability of p53 transcriptional activation domain
is directly related to the binding affinity to TATA-binding protein. _J. Biol. Chem._ 270: 25014–25019. Article CAS PubMed Google Scholar * Zilfou JT, Hoffman WH, Sank M, George DL and
Murphy M (2001) The corepressor mSin3a interacts with the proline-rich domain of p53 and protects p53 from proteasome-mediated degradation. _Mol. Cell. Biol._ 21: 3974–3985. Article CAS
PubMed PubMed Central Google Scholar * Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP and Xiao ZX (2002) The prolyl isomerase Pin1 is a regulator of p53 in
genotoxic response. _Nature_ 419: 849–853. Article CAS PubMed Google Scholar * Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C and Del
Sal G (2002) The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. _Nature_ 419: 853–857. Article CAS PubMed Google Scholar * Dornan D, Shimizu
H, Burch L, Smith AJ and Hupp TR (2003) The proline repeat domain of p53 binds directly to the transcriptional coactivator p300 and allosterically controls DNA-dependent acetylation of p53.
_Mol. Cell. Biol._ 23: 8846–8861. Article CAS PubMed PubMed Central Google Scholar * Curtin JC and Spinella MJ (2005) p53 in human embryonal carcinoma: identification of a transferable,
transcriptional repression domain in the N-terminal region of p53. _Oncogene_ 24: 1481–1490. Article CAS PubMed Google Scholar * Bond GL, Hu W and Levine AJ (2005) MDM2 is a central
node in the p53 pathway: 12 years and counting. _Curr. Cancer Drug Targets_ 5: 3–8. Article CAS PubMed Google Scholar * Zhang Y and Xiong Y (2001) A p53 amino-terminal nuclear export
signal inhibited by DNA damage-induced phosphorylation. _Science_ 292: 1910–1915. Article CAS PubMed Google Scholar * Shieh S-Y, Ikeda M, Taya Y and Prives C (1997) DNA damage-induced
phosphorylation of p53 alleviates inhibition by MDM2. _Cell_ 91: 325–334. Article CAS PubMed Google Scholar * Chehab NH, Malikzay A, Stavridi ES and Halazonetis TD (1999) Phosphorylation
of Ser-20 mediates stabilization of human p53 in response to DNA damage. _Proc. Natl. Acad. Sci. USA_ 96: 13777–13782. Article CAS PubMed PubMed Central Google Scholar * Dumaz N and
Meek DW (1999) Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. _EMBO J._ 18: 7002–7010. Article CAS PubMed PubMed Central
Google Scholar * Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R and Brady JN (1998) Phosphorylation of p53 serine 15 increases interaction with CBP. _J. Biol. Chem._ 273:
33048–33053. CAS PubMed Google Scholar * Saito S, Goodarzi AA, Higashimoto Y, Noda Y, Lees-Miller SP, Appella E and Anderson CW (2002) ATM mediates phosphorylation at multiple p53 sites,
including Ser(46), in response to ionizing radiation. _J. Biol. Chem._ 277: 12491–12494. Article CAS PubMed Google Scholar * Mayo LD, Seo YR, Jackson MW, Smith ML, Rivera Guzman J,
Korgaonkar CK and Donner DB (2005) Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. _J. Biol. Chem._ 280:
25953–25959. Article CAS PubMed Google Scholar * Saito S, Yamaguchi H, Higashimoto Y, Chao C, Xu Y, Fornace Jr AJ, Appella E and Anderson CW (2003) Phosphorylation site interdependence
of human p53 post-translational modifications in response to stress. _J. Biol. Chem._ 278: 37536–37544. Article CAS PubMed Google Scholar * Ashcroft M, Kubbutat MH and Vousden KH (1999)
Regulation of p53 function and stability by phosphorylation. _Mol. Cell Biol._ 19: 1751–1758. Article CAS PubMed PubMed Central Google Scholar * Thompson T, Tovar C, Yang H, Carvajal D,
Vu BT, Xu Q, Wahl GM, Heimbrook DC and Vassilev LT (2004) Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. _J. Biol. Chem._ 279:
53015–53022. Article CAS PubMed Google Scholar * Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, Wang W, Kashanchi F and Shiekhattar R (2000) BRCA1 is associated with a human
SWI/SNF-related complex: linking chromatin remodeling to breast cancer. _Cell_ 102: 257–265. Article CAS PubMed Google Scholar * Lee D, Kim JW, Seo T, Hwang SG, Choi EJ and Choe J (2002)
SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. _J. Biol. Chem._ 277: 22330–22337. Article CAS PubMed Google
Scholar * Hill DA, de la Serna IL, Veal TM and Imbalzano AN (2004) BRCA1 interacts with dominant negative SWI/SNF enzymes without affecting homologous recombination or radiation-induced
gene activation of p21 or Mdm2. _J. Cell Biochem._ 91: 987–998. Article CAS PubMed Google Scholar * Hsu CH, Chang MD, Tai KY, Yang YT, Wang PS, Chen CJ, Wang YH, Lee SC, Wu CW and Juan
LJ (2004) HCMV IE2-mediated inhibition of HAT activity downregulates p53 function. _EMBO J._ 23: 2269–2280. Article CAS PubMed PubMed Central Google Scholar * Barlev NA, Liu L, Chehab
NH, Mansfield K, Harris KG, Halazonetis TD and Berger SL (2001) Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. _Mol. Cell_ 8:
1243–1254. Article CAS PubMed Google Scholar * Lill NL, Grossman SR, Ginsberg D, DeCaprio J and Livingston DM (1997) Binding and modulation of p53 by p300/CBP coactivators. _Nature_ 387:
823–827. Article CAS PubMed Google Scholar * Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS and Kelly K (1997) Recruitment of p300/CBP in p53-dependent signal pathways.
_Cell_ 89: 1175–1184. Article CAS PubMed Google Scholar * An W, Kim J and Roeder RG (2004) Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53.
_Cell_ 117: 735–748. Article CAS PubMed Google Scholar * Gu W, Malik S, Ito M, Yuan CX, Fondell JD, Zhang X, Martinez E, Qin J and Roeder RG (1999) A novel human SRB/MED-containing
cofactor complex, SMCC, involved in transcription regulation. _Mol. Cell_ 3: 97–108. Article CAS PubMed Google Scholar * Zhang X, Krutchinsky A, Fukuda A, Chen W, Yamamura S, Chait BT
and Roeder RG (2005) MED1/TRAP220 exists predominantly in a TRAP/Mediator subpopulation enriched in RNA polymerase II and is required for ER-mediated transcription. _Mol. Cell_ 19: 89–100.
Article CAS PubMed Google Scholar * Xing J, Sheppard HM, Corneillie SI and Liu X (2001) p53 Stimulates TFIID-TFIIA-promoter complex assembly, and p53-T antigen complex inhibits TATA
binding protein-TATA interaction. _Mol. Cell Biol._ 21: 3652–3661. Article CAS PubMed PubMed Central Google Scholar * Graunke DM, Fornace Jr AJ and Pieper RO (1999) Presetting of
chromatin structure and transcription factor binding poise the human GADD45 gene for rapid transcriptional up-regulation. _Nucleic Acids Res._ 27: 3881–3890. Article CAS PubMed PubMed
Central Google Scholar * Xiao G, White D and Bargonetti J (1998) p53 binds to a constitutively nucleosome free region of the mdm2 gene. _Oncogene_ 16: 1171–1181. Article CAS PubMed
Google Scholar * Braastad CD, Han Z and Hendrickson EA (2003) Constitutive DNase I hypersensitivity of p53-regulated promoters. _J. Biol. Chem._ 278: 8261–8268. Article CAS PubMed Google
Scholar * Harms K, Nozell S and Chen X (2004) The common and distinct target genes of the p53 family transcription factors. _Cell. Mol. Life Sci._ 61: 822–842. Article CAS PubMed Google
Scholar * Yu J and Zhang L (2005) The transcriptional targets of p53 in apoptosis control. _Biochem. Biophys. Res. Commun._ 331: 851–858. Article CAS PubMed Google Scholar * Mirza A,
Wu Q, Wang L, McClanahan T, Bishop WR, Gheyas F, Ding W, Hutchins B, Hockenberry T, Kirschmeier P, Greene JR and Liu S (2003) Global transcriptional program of p53 target genes during the
process of apoptosis and cell cycle progression. _Oncogene_ 22: 3645–3654. Article CAS PubMed Google Scholar * Burns TF and El-Deiry WS (2003) Microarray analysis of p53 target gene
expression patterns in the spleen and thymus in response to ionizing radiation. _Cancer Biol. Ther._ 2: 431–443. Article CAS PubMed Google Scholar * Sax JK, Stoddard A, Murphy ME,
Chodosh L and El-Deiry WS (2003) Microarray expression profiling of p53-dependent transcriptional changes in an immortalized mouse embryo fibroblast cell line. _Cancer Biol. Ther._ 2:
416–430. Article CAS PubMed Google Scholar * Robinson M, Jiang P, Cui J, Li J, Wang Y, Swaroop M, Madore S, Lawrence TS and Sun Y (2003) Global genechip profiling to identify genes
responsive to p53-induced growth arrest and apoptosis in human lung carcinoma cells. _Cancer Biol. Ther._ 2: 406–415. Article CAS PubMed Google Scholar * Zhu N, Gu L, Findley HW and Zhou
M (2005) Transcriptional repression of the eukaryotic initiation factor 4E gene by wild type p53. _Biochem. Biophys. Res. Commun._ 335: 1272–1279. Article CAS PubMed Google Scholar *
Kim E, Gunther W, Yoshizato K, Meissner H, Zapf S, Nusing RM, Yamamoto H, Van Meir EG, Deppert W and Giese A (2003) Tumor suppressor p53 inhibits transcriptional activation of invasion gene
thromboxane synthase mediated by the proto-oncogenic factor ets-1. _Oncogene_ 22: 7716–7727. Article CAS PubMed Google Scholar * Maiyar AC, Phu PT, Huang AJ and Firestone GL (1997)
Repression of glucocorticoid receptor transactivation and DNA binding of a glucocorticoid response element within the serum/glucocorticoid-inducible protein kinase (sgk) gene promoter by the
p53 tumor suppressor protein. _Mol. Endocrinol._ 11: 312–329. Article CAS PubMed Google Scholar * Liu G, Schwartz JA and Brooks SC (1999) p53 down-regulates ER-responsive genes by
interfering with the binding of ER to ERE. _Biochem. Biophys. Res. Commun._ 264: 359–364. Article CAS PubMed Google Scholar * Maeda Y, Seidel SD, Wei G, Liu X and Sladek FM (2002)
Repression of hepatocyte nuclear factor 4alpha tumor suppressor p53: involvement of the ligand-binding domain and histone deacetylase activity. _Mol. Endocrinol._ 16: 402–410. CAS PubMed
Google Scholar * Gu L, Zhu N, Findley HW, Woods WG and Zhou M (2004) Identification and characterization of the IKKalpha promoter: positive and negative regulation by ETS-1 and p53,
respectively. _J. Biol. Chem._ 279: 52141–52149. Article CAS PubMed Google Scholar * St Clair S, Giono L, Varmeh-Ziaie S, Resnick-Silverman L, Liu WJ, Padi A, Dastidar J, DaCosta A,
Mattia M and Manfredi JJ (2004) DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. _Mol. Cell_
16: 725–736. Article PubMed Google Scholar * Ori A, Zauberman A, Doitsh G, Paran N, Oren M and Shaul Y (1998) p53 binds and represses the HBV enhancer: an adjacent enhancer element can
reverse the transcription effect of p53. _EMBO J._ 17: 544–553. Article CAS PubMed PubMed Central Google Scholar * Lee KC, Crowe AJ and Barton MC (1999) p53-mediated repression of
alpha-fetoprotein gene expression by specific DNA binding. _Mol. Cell Biol._ 19: 1279–1288. Article CAS PubMed PubMed Central Google Scholar * Li B and Lee MY (2001) Transcriptional
regulation of the human DNA polymerase delta catalytic subunit gene POLD1 by p53 tumor suppressor and Sp1. _J. Biol. Chem._ 276: 29729–29739. Article CAS PubMed Google Scholar *
Subbaramaiah K, Altorki N, Chung WJ, Mestre JR, Sampat A and Dannenberg AJ (1999) Inhibition of cyclooxygenase-2 gene expression by p53. _J. Biol. Chem._ 274: 10911–10915. Article CAS
PubMed Google Scholar * Murphy M, Ahn J, Walker KK, Hoffman WH, Evans RM, Levine AJ and George DL (1999) Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated
by interaction with mSin3a. _Genes Dev._ 13: 2490–2501. Article CAS PubMed PubMed Central Google Scholar * Koumenis C, Alarcon R, Hammond E, Sutphin P, Hoffman W, Murphy M, Derr J,
Taya Y, Lowe SW, Kastan M and Giaccia A (2001) Regulation of p53 by hypoxia: dissociation of transcriptional repression and apoptosis from p53-dependent transactivation. _Mol. Cell Biol._
21: 1297–1310. Article CAS PubMed PubMed Central Google Scholar * Ho JS, Ma W, Mao DY and Benchimol S (2005) p53-Dependent transcriptional repression of c-myc is required for G1 cell
cycle arrest. _Mol. Cell Biol._ 25: 7423–7431. Article CAS PubMed PubMed Central Google Scholar * Johnson RA, Shepard EM and Scotto KW (2005) Differential regulation of MDR1
transcription by the p53 family members. Role of the DNA binding domain. _J. Biol. Chem._ 280: 13213–13219. Article CAS PubMed Google Scholar * Sengupta S, Shimamoto A, Koshiji M, Pedeux
R, Rusin M, Spillare EA, Shen JC, Huang LE, Lindor NM, Furuichi Y and Harris CC (2005) Tumor suppressor p53 represses transcription of RECQ4 helicase. _Oncogene_ 24: 1738–1748. Article CAS
PubMed Google Scholar * Chun AC and Jin DY (2003) Transcriptional regulation of mitotic checkpoint gene MAD1 by p53. _J. Biol. Chem._ 278: 37439–37450. Article CAS PubMed Google
Scholar * Wilkinson DS, Ogden SK, Stratton SA, Piechan JL, Nguyen TT, Smulian GA and Barton MC (2005) A direct intersection between p53 and transforming growth factor beta pathways targets
chromatin modification and transcription repression of the alpha-fetoprotein gene. _Mol. Cell Biol._ 25: 1200–1212. Article CAS PubMed PubMed Central Google Scholar * Rocha S, Martin
AM, Meek DW and Perkins ND (2003) p53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-kappaB subunit with histone
deacetylase 1. _Mol. Cell Biol._ 23: 4713–4727. Article CAS PubMed PubMed Central Google Scholar * Gottifredi V, Karni-Schmidt O, Shieh SS and Prives C (2001) p53 down-regulates CHK1
through p21 and the retinoblastoma protein. _Mol. Cell Biol._ 21: 1066–1076. Article CAS PubMed PubMed Central Google Scholar * Lohr K, Moritz C, Contente A and Dobbelstein M (2003)
p21/CDKN1A mediates negative regulation of transcription by p53. _J. Biol. Chem._ 278: 32507–32516. Article CAS PubMed Google Scholar * Murphy M, Hinman A and Levine AJ (1996) Wild-type
p53 negatively regulates the expression of a microtubule-associated protein. _Genes Dev._ 10: 2971–2980. Article CAS PubMed Google Scholar * Venot C, Maratrat M, Dureuil C, Conseiller E,
Bracco L and Debussche L (1998) The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with
transcriptional repression. _EMBO J._ 17: 4668–4679. Article CAS PubMed PubMed Central Google Scholar * Li J, Tan M, Li L, Pamarthy D, Lawrence TS and Sun Y (2005) SAK, a new polo-like
kinase, is transcriptionally repressed by p53 and induces apoptosis upon RNAi silencing. _Neoplasia_ 7: 312–323. Article CAS PubMed PubMed Central Google Scholar * Gridasova AA and
Henry RW (2005) The p53 tumor suppressor protein represses human snRNA gene transcription by RNA polymerases II and III independently of sequence-specific DNA binding. _Mol. Cell Biol._ 25:
3247–3260. Article CAS PubMed PubMed Central Google Scholar * Zhai W and Comai L (2000) Repression of RNA polymerase I transcription by the tumor suppressor p53. _Mol. Cell Biol._ 20:
5930–5938. Article CAS PubMed PubMed Central Google Scholar * Budde A and Grummt I (1999) p53 represses ribosomal gene transcription. _Oncogene_ 18: 1119–1124. Article CAS PubMed
Google Scholar * Stein T, Crighton D, Warnock LJ, Milner J and White RJ (2002) Several regions of p53 are involved in repression of RNA polymerase III transcription. _Oncogene_ 21:
5540–5547. Article CAS PubMed Google Scholar * Crighton D, Woiwode A, Zhang C, Mandavia N, Morton JP, Warnock LJ, Milner J, White RJ and Johnson DL (2003) p53 represses RNA polymerase
III transcription by targeting TBP and inhibiting promoter occupancy by TFIIIB. _EMBO J._ 22: 2810–2820. Article CAS PubMed PubMed Central Google Scholar * Eichhorn K and Jackson SP
(2001) A role for TAF3B2 in the repression of human RNA polymerase III transcription in nonproliferating cells. _J. Biol. Chem._ 276: 21158–21165. Article CAS PubMed Google Scholar *
Cairns CA and White RJ (1998) p53 is a general repressor of RNA polymerase III transcription. _EMBO J._ 17: 3112–3123. Article CAS PubMed PubMed Central Google Scholar * Bourdon JC,
Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK and Lane DP (2005) p53 isoforms can regulate p53 transcriptional activity. _Genes Dev._ 19: 2122–2137. Article CAS
PubMed PubMed Central Google Scholar * Rohaly G, Chemnitz J, Dehde S, Nunez AM, Heukeshoven J, Deppert W and Dornreiter I (2005) A novel human p53 isoform is an essential element of the
ATR-intra-S phase checkpoint. _Cell_ 122: 21–32. Article CAS PubMed Google Scholar * Harms KL and Chen X (2006) The functional domains in p53 family proteins exhibit both common and
distinct properties. _Cell Death Differ_. 13: 890–897. Article CAS PubMed Google Scholar Download references AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Biological
Sciences, Columbia University, 530 120th Street, New York, 10027, NY, USA O Laptenko & C Prives Authors * O Laptenko View author publications You can also search for this author inPubMed
Google Scholar * C Prives View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to C Prives. ADDITIONAL INFORMATION Edited
by G Melino RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Laptenko, O., Prives, C. Transcriptional regulation by p53: one protein, many possibilities.
_Cell Death Differ_ 13, 951–961 (2006). https://doi.org/10.1038/sj.cdd.4401916 Download citation * Received: 17 January 2006 * Revised: 28 February 2006 * Accepted: 28 February 2006 *
Published: 31 March 2006 * Issue Date: 01 June 2006 * DOI: https://doi.org/10.1038/sj.cdd.4401916 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative
KEYWORDS * p53 * transcriptional activation/suppression * functional domain * co-activator