Physical map and cosmid contig encompassing a new interstitial deletion of the x-linked lymphoproliferative syndrome region
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The X-linked lymphoproliferative syndrome (XLP) is an inherited immunodeficiency to Epstein-Barr virus infection that has been mapped to chromosome Xq25. Molecular analysis of XLP
patients from ten different families identified a small interstitial constitutional deletion in 1 patient (XLP-D). This deletion, initially defined by a single marker, DF83, known to map to
interval Xq24–q26.1, is nested within a previously reported and much larger deletion in another XLP patient (XLP-739). A cosmid minilibrary was constructed from a single mega-YAC and used to
establish a contig encompassing the whole XLP-D deletion and a portion of the XLP-739 deletion. Based on this contig, the size of the XLP-D deletion can be estimated at 130 kb. The
identification of this minimal deletion, within which at least a portion of the XLP gene is likely to reside, should greatly facilitate efforts in isolating the gene. You have full access to
this article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS IDENTIFICATION AND ANALYSIS OF DELETION BREAKPOINTS IN FOUR MOHR-TRANEBJÆRG SYNDROME (MTS) PATIENTS
Article Open access 02 September 2022 EXPERIMENTAL METHOD FOR HAPLOTYPE PHASING ACROSS THE ENTIRE LENGTH OF CHROMOSOME 21 IN TRISOMY 21 CELLS USING A CHROMOSOME ELIMINATION TECHNIQUE Article
Open access 31 May 2022 OPTICAL MAPPING OF THE 22Q11.2DS REGION REVEALS COMPLEX REPEAT STRUCTURES AND PREFERRED LOCATIONS FOR NON-ALLELIC HOMOLOGOUS RECOMBINATION (NAHR) Article Open access
22 July 2020 INTRODUCTION X-linked lymphoproliferative syndrome (XLP; MIM 308240), originally called Duncan disease [1, 2], is a rare primary immunodeficiency characterized by selective
susceptibility to Epstein-Barr virus (EBV). EBV infection of affected boys results in severe or fatal infectious mononucleosis, with survivors developing hypo- or agammaglobulinemia [1].
Malignant B cell non-Hodgkin lymphoma arises in more than 25% of the survivors [3], with the disease usually fatal by age 40 [4]. Males carrying a defective XLP gene are healthy until they
become infected with EBV, suggesting that their immunodeficiency is specific to this virus [5]. EBV is an ubiquitous herpesvirus, with more than 98% of the adult population showing
immunological evidence of past infection. In addition to self-limited infectious mononucleosis, EBV has been directly implicated in the development of Burkitt lymphoma and nasopharyngeal
carcinoma [6]. EBV is also associated with lymphoproliferative syndromes in individuals with genetic or acquired immunodeficiency, including patients with AIDS or organ transplant recipients
who have undergone immunosuppressive regimens [7]. The XLP syndrome, therefore, provides a genetic model for the interaction between an infectious agent and a congenital immunodeficiency,
and the isolation of the responsible gene should provide insights into the normal control of virally induced lymphoproliferation. The XLP locus, named LYP [8], has been assigned by linkage
studies to the interval Xq24–q26.1, proximal to HPRT [9]. This locus shows 1–2% recombination with DXS42 and 0% with DXS37, both of which are centromeric to it [9–12]. The telomeric border
of the XLP locus is defined by DXS100. Multipoint analysis of this region has suggested a genetic map with the following order of markers [10, 11]: _cen_ − DXS11 − 10 cM − DXS37 − 2 cM −
DXS42 − DXS739 − DXS100 − 7 cM − HPRT − _tel._ This order is consistent with the physical map reported by Wu et al. [13]: _cen_ − DXS42, DXS12 − (2,140 kb) − DXS6 − (700 kb) − DXS982 − (340
kb) − DXS739 − (1,120 kb) − DXS75 − (900 kb) − DXS100 − (1,760 kb) − DXS10 − DXS177 − _tel._ Cytogenetic studies of individuals from XLP families led to the identification of a male with a
large constitutional deletion in the Xq25 region [14]. Genomic sequences corresponding to DXS6, DXS739, and DXS100 were absent in this patient. Two additional patients who had deletions of
markers in the Xq25 region were also reported [15, 16]: 1 had deletions of DXS739 and DXS100 and the 2nd a deletion of DXS739. In both cases, high-resolution cytogenetics of lymphoblastoid
cell lines revealed cytogenetically visible deletions, estimated at 6,000 and 2,000–3,000 kb, respectively. Nonetheless, the large size of this candidate region has complicated positional
cloning efforts. Here, we report a novel constitutional deletion of only 130 kb identified in an XLP patient. This new minimal deletion is spanned by a cosmid contig which should greatly
facilitate efforts at cloning the XLP gene. PATIENTS AND METHODS PATIENTS The patients XLP-D, XLP-M, XLP-S, and XLP-H have been described elsewhere [17]. Patients LB8001, LB8002, and LB8005
are previously undescribed XLP patients, while individuals _XLP-192_, _XLP-724, XLP-739_, and _XLP-978_ are XLP patients studied in our laboratory: _XLP-192_ (corresponding to the IARC192
cell line) was the 3rd son of a French family in which 2 boys had died from lymphoproliferative disease after EBV infection. At the age of 2, he developed fatal infectious mononucleosis with
strong lymphoproliferative syndrome. There was serological evidence of EBV infection in this child. _XLP-724_ (corresponding to the IARC724 cell line) was a 15-year-old Italian boy who
presented with agammaglobulinemia, with no proof of EBV infection. Two of his brothers died at 1 year of age from lymphoproliferative disease. _XLP-739_ (corresponding to the IARC739 cell
line) was a 3-year-old Italian boy who died from lymphoproliferative disease after EBV infection. _XLP-978_ (corresponding to the IARC978 cell line) was a 2-year-old French boy with typical
XLP symptoms, including polyclonal B cell lymphoproliferative disease, hypogammaglobulinemia, and hepatomegaly with abnormal liver functions tests. Immunofluorescence detected that the vast
majority of blood cells expressed EBNA (nuclear antigen) antibodies. The boy died 3 weeks after his symptoms appeared. MARKERS USED The markers used for physical mapping are described in
table 1 [see also refs. 12, 13, 18–20]. DF83, PF12, and CF1a were isolated from a library of human brain cDNAs after screening with sequences generated by laser microdissection of the
terminal portion of the human Xq. These markers were assigned to Xq24–q26.1 [19]. They are considered to be untranslated cDNA and have several stop codons in all reading frames [19]. For
Southern blot analysis, a DF83 subfragment of 400 bp was obtained by polymerase chain reaction (PCR) using the following primers under standard conditions: * DF83a: AGGAATGCACCTACATATGTGGCA
* DF83b: CAGACTGCTTATTTATGGAGATCC Amplification of probes 1D10T3 and 1D10T7 was performed under standard conditions using the following primers: * For 1D10T3: * 1D10T3s:
5′-TTCAAATATAAATCACCTAATAA-3′ * 1D10T3as: 5′-CCAATTGTCTCCTTTTCCAAA-3′ * For 1D10T7: * 1D10T7s: 5′-GCAAGGGAATCTATGGAACTG-3′ * 1D10T7as: 5′-GGTGCAACTAATTAGTTGGGC-3′ H3-4 (DXS739) is a 1.5-kb
_Hind_III fragment from phage bA6. [Nelson, personal commun.; 13]. The preparation of riboprobes used for cosmid walking is described in the section Cosmid Isolation and Construction of
Contigs. SCREENING OF THE YAC (YEAST ARTIFICIAL CHROMOSOME) LIBRARY YAC 916D1 was isolated by PCR screening of pooled DNAs of the CEPH mega-YAC library using oligos from X2-183 (DXS982). YAC
yWXD592 was obtained from the Center for Genetics in Medicine (St. Louis, Mo., USA) and was localized within large XLP deletions previously described [14]. Yeast cell culture and DNA
isolation were performed as described [21]. _PCR Conditions and Primers._ PCR was performed in 50 µl of the reaction mixture for 40 cycles using a one-step cycle program of 94°C for 4 min,
55 for 2, and 72°C for 2 min and an additional extension of 10 min at 72°C. The X2-183 primers (GDB ID: G00-216-816) used were: * TA12T7: * 5′-CCGTCACATATAGAGATAGTATTACTGAACC-3′ * TA12SP6: *
5′-GGTCATTATGAATGGTCAAAGGAATTTGACTGC-3′ The amplified products (1.1 kb) were electrophoresed in 1.1% agarose. YAC 916D1 COSMID MINILIBRARY AND GRIDDED X CHROMOSOME LIBRARY For construction
of cosmid libraries from YAC 916D1 in the supercos vector, the following procedure was used: 2 µg of genomic DNA from a yeast strain carrying YAC 916D1 was prepared in agarose blocks,
partially digested with 0.1 U of _Mbo_I (New England Biolabs, Beverly, Mass., USA), and dephosphorylated with 0.1 U calf intestinal phosphatase (New Englands Biolabs). Yeast DNA was then
ligated into the _BAM_HI site of Supercos vector (Stratagene, La Jolla, Calif., USA), previously linearized with _Xba_I and dephosphorylated, and packaged (Stratagene). XLI Blue MR cells
(Stratagene) grown in 0.2% maltose and 10 m_M_ MgSO4 were infected with packaged DNA and selected for growth in ampicillin. Colonies containing human-derived genomic DNA were identified by
hybridization to 32P-labeled Cot-1 DNA and arrayed in 96-well plates. The ICRF-gridded X chromosome flow-sorted cosmid library is a reference library in Lawrist 4 vector [22]. The sources of
cosmids used for physical mapping are listed below. Cosmids from ICRF were (the official reference name is in parentheses): * 1818 (ICRFc104E1818), 64 (ICRFc104J201), * E079 (ICRFc104E079),
B0920 (ICRFc104B0920), * P1213 (ICRFc104P1213Q), L0129 (ICRFc104L0129), * L2015 (ICRFc104L015QD), N1335 (ICRFc104N1335), * D1785 (ICRFcl04N1785), D8112 (ICRFc104D8112), * 4 (ICRFc104D196),
31 (ICRFc104D1912), and * 100 (ICRFc104H07100). Cosmids from the YAC 916D1 minilibrary were: 1D10, E8, C8, 1N5, 1U8, A8, 1Z4, F11, 1X6, 1J11, A12, and 1H11. COSMID ISOLATION AND CONSTRUCTION
OF CONTIGS DNAs from individual cosmids of the YAC 916D1 cosmid library were prepared by classical alkaline lysis DNA extraction [23] and spotted onto nylon filters by manual dot blot. Dot
blot filters were hybridized with probes mapped in the Xq25 region. ICRF-gridded X chromosome cosmid filters were hybridized with the probes as described by Nizetic et al. [22]. DNA was
extracted from hybridized clones by alkaline lysis as previously described [23]. Cosmid clones were used as a starting point for chromosome walking. For walking, terminal-end riboprobes of
cosmid and P1 clones were synthesized and hybridized to cosmid dot blots. Positive clones were selected, and the digested DNA from these clones was compared with that of flanking cosmids,
blotted, and probed with the riboprobe originally used to select the cosmid from dot blots. Riboprobes were synthesized as follows: 3 µg of cosmid DNA was digested by _Rsa_I (Promega,
Madison, Wisc., USA), extracted with phenol-chloroform, and ethanol precipitated. The reactions were performed in 25 µl final volume with 1 m_M_ nucleotide (ACG), 30 mM dithiothreitol, and 5
µCi 32P-UTP using 10 U of RNA polymerase at 37 ° C for 30 min. T3 and T7 RNA polymerases were used for YAC 916D1 cosmid library (in supercos vector), SP6, and T7 RNA pol. For ICRF cosmid
library (in Lawrist 4) 10 U of RNAse-free DNAse was then added to the reaction mixture and incubated for 15 min at 37°C. Finally, riboprobes were phenol-chloroform extracted and ethanol
precipitated before hybridization. This process of terminal-end riboprobes and filter hybridization of cosmid DNAs was repeated at each stage of the walk. Cosmid overlaps were verified by
the DNA fingerprinting. Cosmid DNAs were _Hinf_I or _Taq_I digested, blotted, and probed with total human placental DNA. Cosmids were subcloned into pBluescript (Stratagene) using standard
techniques [23]. RESTRICTION MAP A restriction map with _Bam_HI was generated by partial digestion of cosmid DNA and subsequently confirmed by complete digestion, while the map of a rare
cutter (_Cla_I) was generated only by complete digestion. For the partial digestion approach, cosmids cloned in Lawrist 4 vector were linearized with _Eag_I (position 4272 of the vector).
Partially digested DNA fragments were visualized by hybridization with two end probes, Law22 (nucleotides 5027–5281 of the vector) and Law33 (nucleotides 3870–4212), respectively, and
amplified from the vector by PCR in standard conditions. The primer sequences for amplifying these two probes were: * Law22a: TTCTGAGCGGGACTCTGG * Law22b: TGCACTGCCGGTAGAACTC * Law33a:
CAAGCTAGCTTCACGCTGC * Law33b: TGATCAGATCTTGATCCCCTG Cosmid clones in Supercos vector were linearized with _Bss_HII (position 4256 of the vector). The two end probes were Sup 43-290
(nucleotides 4395–4684 of the vector) and Sup 38-270 (nucleotides 3899–4168). Primer sequences were: * Sup 43-290a: CGTTGGCTACCCGTGATATT * Sup 43-290b: ACTCCAGCATGAGATCCCC * Sup 38-270b:
CTGAATGAACTGCAGGACGA * Sup 38-270b: AGTACGTGCTCGCTCGATG SOUTHERN BLOT ANALYSIS Isolation of high-molecular-weight DNA, restriction endonuclease digestion, DNA separation and blotting, and
dotting of DNA fragments were performed as described previously [23]. Cosmid inserts were hybridized in the presence of excess human competitor DNA as previously described [24]. The filters
were hybridized in Church solution − 0.5 _M_ phosphate buffer, pH 7.2, 7% sodium dodecyl sulfate (SDS), 1% bovine serum albumin — at 65°C and then washed once stringently in 2 × SSC and 0.1%
SDS at 65°C before washing in 0.1 × SSC and 0.1% SDS at 65°C (1 × SSC = 0.15 _M_ NaCl, 1.5 _M_ sodium citrate). DNA probes were 32P-labeled by random priming (Amersham) and used at a
specific activity of 1 million cpm/ml of hybridization solution. Riboprobes were hybridized under the same conditions in Church buffer. RESULTS IDENTIFICATION OF AN XLP PATIENT CARRYING A
SMALL DELETION To define a minimal interval containing the XLP gene, EBV-immortalized cell lines were generated from 10 unrelated male patients. These were screened for hemizygous deletion
or rearrangement of Xq25 markers (table 2). Markers known to be deleted in previous patients [14–16], X2-183 (DXS982), H3-4 (DXS739), and p45-h (DXS100), were tested. In addition, three new
markers, recently mapped to Xq25-26, CF1a, DF83, and PF12 [19], were found to be deleted in patient XLP-739 who has a large deletion, spanning at least 2,000 kb, and, therefore, localized in
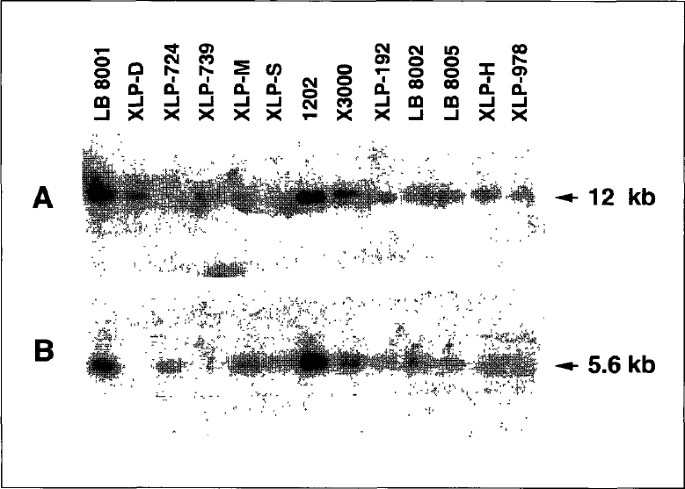
the XLP region locus. Screening patients by both Southern blot [25] and/or PCR amplification identified a patient, XLP-D, lacking only one marker: DF83 (fig. 1). This patient was,
therefore, presumed to carry a small interstitial deletion that might define the smallest region of overlap with the deletion of patient XLP-739. ISOLATION OF YAC CLONES FROM THE DELETED
REGION The CEPH megayac library was screened with probes X2-183 (DXS982) and DF83. The YAC 916D1 (1.74 Mbp) was found to contain X2-183 and DF83. Another YAC previously identified in the
Xq25 region (yWXD592) also contained the same two markers. YAC916D1 was characterized and compared with yWXD592 using additional markers — PF12, H3-4 (DXS739), CFla, and p45h (DXS100) — with
the following results: (1) YAC 916D1 was found to contain probes PF12, CFla, and H3-4 (DXS739), but not p45h (DXS100), and (2) YAC yWXD592 was found to be negative for these four markers.
The pattern of overlap among these markers indicates that DF83, X2-183 (DXS982), and PF12 lie relatively close to each other. YAC 916D1 and yWXD592 contain both DF83 which is deleted in
patient XLP-D and X2-183 which is not deleted in this patient, indicating that these YACs contain at least one breaking point of the XLP-D deletion. Since DXS982 maps centromeric to DXS739
[13], the minimal deletion of XLP-D lies centromeric to DXS982 and DXS739 (fig. 2). CONSTRUCTION OF COSMID CONTIG A cosmid library was generated from YAC 916D1 which appeared to span the
XLP-D deletion. Whole-yeast DNA was partially digested with Sau3A and cloned into the SuperCos vector (Stratagene). Recombinants were screened for the presence of human repetitive sequences
(Cotl), and 400 cosmids were arrayed, representing an estimated threefold coverage of YAC 916D1. Additional clones were obtained from the Imperial Cancer Research Fund gridded human X
chromosome cosmid library [22]. Four cosmids positive for DF83, one cosmid containing X2-183 (DXS982), two cosmids containing PF12, and one cosmid containing H3-4 (DXS739) were isolated and
used as a starting point for chromosome walking. End probes from the T3 or T7 ends of cosmids were generated either as riboprobes or by sequencing followed by PCR amplification of end
fragments. At each step of the chromosome walk, cosmids fragments were tested by Southern blotting to confirm their presence or absence within the deleted region. Four initial contigs around
markers DF83, X2-183 (DXS982), PF12, and H3-4 (DXS739) were linked to form a contig with a gap in it partially encompassing the deletion of patient XLP-739 and completely covering the
deletion of patient XLP-D. Since cosmids from the contig linking DXS739 and DXS982 do not contain probes deleted in patient XLP-D, we can localize the XLP-D deletion centromeric to DXS982. A
gap was not filled in between the XLP-D deletion and marker X2-183 (DXS982; fig. 2), even after several screening efforts of the X chromosome cosmid library. The size of this gap remains,
therefore, unknown. Overlap between cosmids in the contig was verified by DNA fingerprinting using repetitive sequence probes. A restriction map based on one frequent-cutter enzyme (_Bam_HI)
and one rare-cutter enzyme (_Cla_I) was constructed for the centromeric high-density part of the contig covering the smaller deletion (figure 3). This restriction map makes it possible to
estimate precisely the length of overlap between different cosmids. CHARACTERIZATION OF BREAKING POINTS To characterize the two breaking points of the XLP-D deletion, we hybridized _Eco_RI
and _Bam_HI subfragments of three cosmids containing deleted and nondeleted sequences (1D10, E079, and B0920) on genomic DNA from patient XLP-D and normal individuals digested with several
restriction enzymes (_Eco_RI, _Hind_III_-Bam_HI, _Pst_1). By Southern blot analysis, a nondeleted probe from cosmid ID 10 identified in patient XLP-D a larger fragment with respect to normal
controls after _Hind_III digestion (fig. 4). Sixty normal individuals were tested to exclude the possibility of polymorphisms. The breaking point was finally localized in a 7-kb
_Hind_III_-Bam_HI fragment of 1D10. The other breaking point was mapped in a 10-kb region covered by cosmids E079 and B0920 and delimited by probes JL17 (deleted in XLP-D) and B0920.7
(nondeleted in XLP-D; see figure 4). It was not possible to identify in this 10-kb region, which is rich in repetitive sequences, a single-copy probe which could be used to localize more
precisely the breaking point. The restriction map of this region (not shown) was found to be identical in both cosmids, excluding the possibility of chimerism. Based on the data just
summarized, the length of the XLP-D deletion was estimated to be approximately 130 kb. DISCUSSION We identified a new XLP patient harboring the smallest deletion in the XLP region reported
so far (estimated at approximately 130 kb). A cosmid contig covering 500 kb of genomic DNA and encompassing completely this new deletion was constructed. A physical linkage between several
markers in the region was obtained, and new markers were localized precisely with respect to previously known markers in Xq25. In particular, markers that had been assigned to Xq24–q26.1
(DF83, PF12 and CF1a) by use of somatic cell hybrid panels [19] were physically linked in a 1.5-Mbp region of Xq25 contained in YAC 916D1. Moreover, DF83 was mapped as centromeric to PF12
[19]. The position of CF1a with respect to the contig is unknown, as no other YAC, with the exception of YAC 916D1, nor cosmid was found to contain this probe. The physical map presented
here adds useful information about polymorphic markers of this region. DXS982 is physically linked to the XLP-D deletion as indicated by YAC yWXD592. DXS739 and PF12 are very close (50 kb),
and the two markers DXS982 and DXS739 are physically linked. As DXS982 is centromeric to DXS739, the following sequence of markers can be drawn: * _cen_ − del XLP-D − DXS982 − PF12 − DXS739
− _tel._ In their high-resolution map of the region, Wu et al. [13] studied the relative position of a set of markers from Xq24–q26.1 using fluorescence in situ hybridization. These authors
estimated the distance between DXS982 and DXS739 at 340 kb with an uncertainty of 70 kb. This estimate is in agreement with the distance of 300 kb we obtained using our cosmid contig. A gap,
covered by YACs yWXD592 and 916D1, is not represented in our cosmids. Efforts to isolate further cosmids between cosmids P1213 and L0129 were unsuccessful. Because of this gap, we are
unable to give an orientation towards the centromeric cosmid contig covering the XLP-D deletion. Because of the limited number and the small size of XLP families, it was very difficult to
restrict the critical XLP region by genetic analysis, and the XLP gene could be localized only in a 2-cM interval between DXS42 and DXS100. The identification of XLP patients harboring
interstitial deletions represented an important step in reducing this interval. Studies using fluorescence in situ hybridization [13] and physical mapping with YACs [26] assigned the XLP
gene to a about a 2-Mb interval between DXS6 and DXS100, around DXS739, which is deleted in 3 XLP patients [15]. The identification of the new interstitial deletion in XLP-D, therefore,
reduces considerably the candidate region for the XLP gene. The XLP-D deletion which maps in the previously defined large candidate region is physically linked to DXS982 and is centromeric
to DXS739. The restriction map of the region deleted in patient XLP-D will be instrumental in the positional cloning of the gene. Single-copy fragments from cosmids within the minimal XLP
deletion contig were used as probes to search for small genomic rearrangements in our panel of XLP patients. No new abnormal fragments were seen by Southern blotting, indicating that the
XLP-D deletion defines the smallest genomic interval within which a fragment of the XLP gene must reside. The new markers that we have assigned to the XLP locus, DF83 and PF12, are
potentially interesting, since they were derived from expressed sequences [19]. However, they do not contain an open reading frame, nor is their expression detectable other than in minimal
amounts by reverse-transcriptase-PCR in all the tissues we have screened. Screening of cDNA libraries with these markers has not identified a further transcribed sequence. Thus, the
relationship, if any, of these transcribed sequences to the XLP gene remains unknown. In the cosmid contig presented here, no clear CpG island was found (data not shown). This finding is in
agreement with the low density of genes in this portion of the X chromosome and also with the possibility that the XLP gene is spread over a large genomic region. The search for conserved
and transcribed sequences in the contig by zoo blot and Northern blot analysis has so far been unsuccessful. As it is not known which cell types are involved in XLP, it is important to study
the expression of candidate sequences in a wide range of tissues. Cells of the immune system, like B and T cells and macrophages, are good candidates for tissue-specific expression of the
XLP gene. Finally, the characterization of the function of the XLP gene should help in understanding the fundamental aspects of EBV biology and the role of the XLP gene in controlling this
viral infection. REFERENCES * Purtilo DT, Yang JP, Cassel CK, Harper P, Stephenson SR, Landin BH, Vawter GF: X-linked recessive progressive combined variable immunodeficiency (Duncan’s
disease). Lancet 1975;i:935–940 Article Google Scholar * On-Line Mendelian Inheritance in Man: OMIM (TM), Immunodeficiency X-Linked Progressive Combined Variable [Lymphoproliferative
Disease, X-linked, XLP], MIM No. 312060. Baltimore, Johns Hopkins University, 1988. * Harrington DS, Weisenburger DD, Purtilo DT: Malignant lymphoma in the X-linked lymphoproliferative
Syndrome. Cancer 1987;59:1419–1429 Article CAS Google Scholar * Purtilo DT, Sakamoto K, Bernabei V, Seeley J, Bechtold T, Rogers G, XLP Collaborators: Epstein-Barr virus induced in boys
with the X-linked lymphoproliferative syndrome (XLP). Update on studies of the registry. Am J Med 1982;73:49–56 Article CAS Google Scholar * Grierson H, Skare J, Hawk J, Panza M, Purtilo
D: Immunoglobulin class and subclass deficiencies prior to EBV infection in males with X-linked lymphoproliferative disease (XLP). Am J Hum Genet 1991;40:294–297 Article CAS Google Scholar
* Miller G: Epstein-Barr virus: Biology, pathogenesis and medical aspects, 1921–1958; in Fields BN, Knipe DM (eds): Virology. New York, Raven Press, 1990. Google Scholar * Klein G:
Epstein-Barr virus strategy in normal and neoplastic B cells. Cell 1994;77:791–793 Article CAS Google Scholar * Mandel JL, Willard HF, Nussbaum RL, Romeo G, Puck JM, Davies KE: Report of
the Committee on the Genetic Constitution of the X Chromosome. Cytogenet Cell Genet 1989;51:384–437 Article CAS Google Scholar * Skare JC, Grierson HL, Sullivan JL, Nussbaum RL, Purtilo
DT, Sylla BS, Lenoir GM, Reilly DS, White BN, Milunsky A: Linkage analysis of seven kindred with the X-linked lymphoproliferative syndrome (XLP) confirms that XLP locus in near DXS42 and
DXS37. Hum Genet 1989;82:354–358 CAS Google Scholar * Sylla BS, Wang Q, Hayoz D, Lathrop GM, Lenoir GM: Multipoint linkage mapping of the Xq25–26 region in a family affected by the
X-linked lymphoproliferative syndrome. Clin Genet 1989;36:459–462 Article CAS Google Scholar * Skare JC, Grierson H, Wyandt H, Sanger W, Milunsky J, Purtilo D, Sullivan J, Milunsky A:
Genetic of the X-linked lymphoproliferative syndrome (abstract 630). Am J Hum Genet 1989;45:A161. Google Scholar * Purtilo DT, Grierson HL: Methods of detection of new families with X
linked lymphoproliferative disease (XLP). Cancer Genet Cytogenet 1991;51:143–153 Article CAS Google Scholar * Wu BL, Milunsky A, Nelson D, Schmeckpeper BJ, Porta G, Schlessinger D, Skare
J: High-resolution mapping of probes near the X-linked lymphoproliferative disease (XLP) locus. Genomics 1993;17:163–170 Article CAS Google Scholar * Wyandt H, Grierson H, Sanger W, Skare
J, Milunsky A, Purtilo D: Chromosome deletion of Xq25 in an individual with X-linked lymphoproliferative disease. Am J Med Genet 1989;33:426–430 Article CAS Google Scholar * Skare J, Wu
BL, Madan S, Pulijaal V, Purtilo D, Haber D, Nelson D, Sylla B, Grierson H, Nitowsky H, Glaser J, Wissink J, White B, Holden J, Housman D, Lenoir G, Wyandt H: Characterization of three
overlapping deletions causing X-linked lymphoproliferative disease. Genomics 1993;16:254–255 Article CAS Google Scholar * Sylla BS, Lamartine J, Wang Q, Pauly S, Lenoir GM, Haber D, Skare
J, Nelson DL, Romeo G, Tsuji O: Characterization of an XLP patient carrying an interstitial deletion at Xq25. Cytogenet Cell Genet 1993;64:189. Article Google Scholar * Purtilo DT,
Sakamoto K, Barnabei V, Seeley J, Bechtold T, Rogers G, Yetz J, Harada S: Epstein-Barr virus-induced diseases in boys with the X-linked lymphoproliferative syndrome (XLP): Update on studies
of the registry. Am J Med 1982;73:49–56 Article CAS Google Scholar * Aldrige J, Kunkel L, Bruns G, Tantravahi U, Lalande M, Brewster T, Moreau E, Wilson M, Bromley W, Roderick T, Latt SA:
A strategy to reveal high frequency RFLPs along the human X chromosome. Am J Hum Genet 1984;36:546–564 Google Scholar * Yokoi H, Hadano S, Kogi M, Kang X, Wakasa K, Ikeda J: Isolation of
expressed sequences encoded by the human Xq terminal portion using microclone probes generated by laser microdissection. Genomics 1994;20:404–411 Article CAS Google Scholar * Mulligan L,
Phillips M, Foster-Gibson C, Beckett J, Partington M, Simpson N, Holden JJ, White BN: Genetic mapping of DNA segments relative to the locus for the fragile-X syndrome at Xq27-3. Am J Hum
Genet 1985;37:463–472 CAS PubMed PubMed Central Google Scholar * Green ED, Olson MV: Systematic screening of yeast artificial chromosome libraries by use of the polymerase chain
reaction. Proc Natl Acad Sci USA 1990;87:1213–1217 Article CAS Google Scholar * Nizetic D, Zehetner G, Monaco AP, Gellen L, Young BD, Lehrach H: Construction, arraying, and high density
screening of large inserts libraries of human chromosome X and 21: Their potential use as reference libraries. Proc Natl Acad Sci USA 1991;88:3233–3237 Article CAS Google Scholar *
Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning. Cold Spring Harbor, Cold Spring Harbor Laboratory, 1989. * Blonden LAJ, den Dunnen JT, van Paassen HMB, Wapenaar MC, Grootscholten PM,
Ginjaar HB, Bakker E, Pearson PL, van Ommen GJB: High resolution deletion breakpoint mapping in the DMD gene by whole cosmid hybridization. Nucleic Acids Res 1989;17:5611–5621 Article CAS
Google Scholar * Kaneko K, Myatake T, Mijeon BR, Warren ST, Tsuji S: Isolation of cosmid clones containing CpG island at Xq24-qter region. Am J Hum Genet 1992;49:2069. Google Scholar *
Wang Q, Ishikawa-Brush Y, Monaco AP, Nelson DL, Caskey CT, Pauly SP, Lenoir GM, Sylla BS: Physical mapping of Xq24–q25 around loci closely linked to the X-linked lymphoproliferative syndrome
locus: An overlapping YAC map and linkage between DXS12, DXS42 and DXS37. Eur J Hum Genet 1993;1:64–71 Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Dr. A.
Harris for providing cell lines. We thank also Dr. D. LePallier for making accessible to us the CEPH mega-YAC library. We thank the ICRF for providing us filters for X chromosome cosmid
library. The technical help of S. Pauly, M.-F. Lavoué, and C. Bonnardel is gratefully acknowledged. We also thank Dr. S. Jones for editing the manuscript. This work was supported in part by
the Ligue Nationale Française contre le Cancer, Comité Départemental du Rhône. F. Heitzmann was supported by a fellowship from the Ligue Nationale Française contre le Cancer, Comité
Départemental de l’Yonne. D. Haber and K. Nichols were supported by grants from NIH (CA4498 for D.H. and NIH CA4509 for K.N.). M. Krainer was supported by a grant from
Erwin-Schrödinger-Stipendium, Fonds zur Förderung der Wissenschaftlichen Forschung. G. Porta was funded by the Associazione Italiana Ricerca sul Cancro and the Telethon e 294. AUTHOR
INFORMATION AUTHORS AND AFFILIATIONS * International Agency for Research on Cancer, 150, cours A. Thomas, F-69372, Lyon Cedex 08, France Jérôme Lamartine, Luo Yin, Fabrice Heitzmann, Simona
Gaudi, Gilbert M. Lenoir, Giovanni Romeo & Bakary S. Sylla PhD * Massachusetts General Hospital Cancer Center and Harvard Medical School, Charlestown, Mass., USA Kim E. Nichols, Michael
Krainer, Amy Bernard, Daniel A. Haber & D. Paul Harkin * Department of Pediatrics, University of Massachusetts Medical Center, Worcester, Mass., USA John L. Sullivan * Institute of
Medical Science, Tokay University School of Medicine, Kanagawa, Japan Joh E. Ikeda * Laboratorio di Genetica Medica, Ospedale San Paolo, Milano, Italia Giovanni Porta * Department of
Genetics, Center for Genetics in Medicine, Washington University School of Medicine, St. Louis, Mo., USA David Schlessinger Authors * Jérôme Lamartine View author publications You can also
search for this author inPubMed Google Scholar * Kim E. Nichols View author publications You can also search for this author inPubMed Google Scholar * Luo Yin View author publications You
can also search for this author inPubMed Google Scholar * Michael Krainer View author publications You can also search for this author inPubMed Google Scholar * Fabrice Heitzmann View author
publications You can also search for this author inPubMed Google Scholar * Amy Bernard View author publications You can also search for this author inPubMed Google Scholar * Simona Gaudi
View author publications You can also search for this author inPubMed Google Scholar * Gilbert M. Lenoir View author publications You can also search for this author inPubMed Google Scholar
* John L. Sullivan View author publications You can also search for this author inPubMed Google Scholar * Joh E. Ikeda View author publications You can also search for this author inPubMed
Google Scholar * Giovanni Porta View author publications You can also search for this author inPubMed Google Scholar * David Schlessinger View author publications You can also search for
this author inPubMed Google Scholar * Giovanni Romeo View author publications You can also search for this author inPubMed Google Scholar * Daniel A. Haber View author publications You can
also search for this author inPubMed Google Scholar * Bakary S. Sylla PhD View author publications You can also search for this author inPubMed Google Scholar * D. Paul Harkin View author
publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Bakary S. Sylla PhD. ADDITIONAL INFORMATION The first two authors contributed
equally to this work. RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Lamartine, J., Nichols, K.E., Yin, L. _et al._ Physical Map and Cosmid Contig
Encompassing a New Interstitial Deletion of the X-Linked Lymphoproliferative Syndrome Region. _Eur J Hum Genet_ 4, 342–351 (1996). https://doi.org/10.1159/000472230 Download citation *
Received: 06 August 1996 * Revised: 08 October 1996 * Accepted: 23 October 1996 * Issue Date: November 1996 * DOI: https://doi.org/10.1159/000472230 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative KEY WORDS * Cosmid contig * Interstitial deletions * X-linked lymphoproliferative syndrome